基于纳米碳点的肿瘤血管抑制剂DX1002特异性定量方法构建
2021-04-24刘书瑶付晓芸雍智全张传维钟卓伶徐小平
刘书瑶,付晓芸,雍智全,张传维,钟卓伶,黄 骏,徐小平,杨 明
(1.四川大学华西药学院,四川 成都 610041; 2.广州安好医药科技有限公司,广东 广州 510700; 3.成都科美迪检测检验有限公司,四川 成都 610041; 4.四川体育职业学院康复科研中心,四川 成都 610041)
0 引 言
碳点(CDs)是一种自带荧光的新型零维纳米聚合材料。因其原料易得、合成简便、安全低毒、生物相容性良好、理化性质和光学性质稳定[1-4]而备受人们关注,被广泛应用于活体成像、生化传感、分析检测、基因治疗[5-8]等领域。DX1002为新型肿瘤血管抑制剂,主要作用于肿瘤新生血管内皮细胞微管蛋白,干扰细胞有丝分裂,产生秋水仙碱样效应,阻塞肿瘤血管,使肿瘤“断养”饿死,目前处临床试验阶段。建立高通量、高灵敏度、特异性的体内分析方法是分析人员一直追求的目标,而面对生物样本存在的数量多、浓度低、杂质干扰严重等难点,人们常采用色谱法(HPLC、GC)或色谱-质谱联用法。其中色谱法可避免大多数杂质干扰而实现良好分离,但存在的前处理繁琐、分析耗时长、灵敏度低等缺陷限制了应用[9-10]。色质联用的出现与普及大大优化了分析时间和分辨率,然而基质效应及对样品预处理的高要求及长分析时间是这一方法的瑕疵[11-13]。要实现高通量分析,光谱法具有较为理想的检测形式,而避免杂质干扰是一个亟待跨越的瓶颈。如今,碳点独有的荧光性、抗漂白性,使其与分析对象间存在唯一性、特异性关系。若供试品与碳点荧光变化量间存在定量关系,则可能构建出适用于药代动力学、刑侦检测等灵敏度、特异性良好的高通量体内检测方法。基于荧光碳量子点的特性,本文拟优化制备出与DX1002存在定量猝灭关系的荧光碳点,建立并验证柠檬酸尿素碳点(CACCDs)探针与DX1002的体外定量关系,为DX1002高通量特异性体内分析方法的建立提供体外定量的依据。
1 实验部分
1.1 仪器与试剂
RF-6000荧光分光光度计(日本岛津分析仪器公司);UV-2300紫外-可见分光光度计(上海天美科学仪器有限公司);LC-2010 HPLC仪(日本岛津分析仪器公司);Sartorios BT 125D十万分之一电子天平(赛多利斯科学仪器(北京)有限公司);SHANGPING FA2004万分之一电子天平(上海天平仪器厂);KL 10260D型超声清洗器(上海荆和分析仪器有限公司);净水机(成都品成科技有限公司);902-ULTS酶标仪(Thermo Fisher);DHG-9电热恒温鼓风干燥箱(上海精宏实验设备有限公司);ZF-I型三用紫外分析仪(上海顾村电光仪器厂);PHS-2F pH计(上海仪电科学仪器股份有限公司);马弗炉(4-10箱式电阻炉,沈阳市节能电炉厂);聚四氟乙烯内衬高压反应釜KH型(上海予申仪器有限公司);RE-52系列旋转蒸发仪(上海雅荣生化设备仪器有限公司);冷冻干燥机(无锡沃鑫信仪器制造有限公司);85-2型恒温磁力搅拌器(上海思乐仪器有限公司)。
柠檬酸(分析纯,天津市科密欧化学试剂有限公司);尿素(分析纯,湖南省芙蓉联合制药厂);罗丹明6G(分析纯,上海麦克林生化科技有限公司);硫酸奎宁(98%,上海麦克林生化科技有限公司)。
1.2 实验方法
1.2.1 柠檬酸尿素碳点(CACCDs)的制备
称取1 g柠檬酸、1 g尿素,研磨混匀,混合粉末置于坩埚中,240 ℃加热4 h,自然冷至室温,加水150 mL,超声溶解,过滤,得CACCDs溶液。
1.2.2 CACCDs的表征
取一定浓度的CACCDs溶液一滴,滴于铜网自然晾干后,用透射电镜(TEM)观察其形貌、粒径,得到透射电镜图。取冷冻干燥的CACCDs粉末与溴化钾粉末,一定比例混合均匀后压制成透明薄片,红外光谱仪测定红外吸收光谱。取一定浓度的CACCDs溶液置于石英比色皿和荧光比色皿中,分别得紫外吸收光谱和荧光光谱。
1.2.3 DX1002的检测
1)对照品溶液的制备
取DX1002对照品250 mg,精密称定,置250 mL棕色容量瓶内,超纯水溶解并稀释至刻度,摇匀,即得每毫升含1 mg DX1002对照品溶液,作储备液,备用。
2)供试品溶液的制备
取 DX1002(20181201)原料药细粉 250 mg,精密称定,置250 mL棕色容量瓶内,超纯水溶解并稀释至刻度,摇匀,作为DX1002供试品溶液,备用。
3)DX1002的测定
依次精密量取CACCDs溶液(1 mg/mL)、上述DX1002溶液各50 μL于0.5 mL的EP管中,再精密加入超纯水100 μL,混匀,转移至96孔板,将加样后的96孔板置酶标仪中测定各孔荧光强度。荧光分光光度法条件为:激发波长(λex):400 nm,发射波长(λem):530 nm,狭缝宽度为12 nm。
2 结果与分析
2.1 CACCDs的工艺优化
经正交实验考察加热时间、加热温度、投料比3个条件,结果如表1所示,当n(柠檬酸∶尿素)=1∶2,于250 ℃下加热240 min时,合成产率及所得CACCDs量子产率最高,分别为26.00%和30.75%。确定了CACCDs最佳合成条件。

表 1 实验因素考察
2.2 CACCDs的表征
图1为CACCDs的表征图,可以看出,CACCDs在透射电镜下呈单分散的类球形颗粒,粒径均一。由CACCDs在红外吸收光谱的。3 301.58 cm-1处吸收峰归属于O-H和N-H的伸缩振动,1 055.83 cm-1处峰属C-N伸缩振动,1 573.62 cm-1处峰属N-H面内弯曲振动,1 481.30 cm-1处峰属C-O弯曲振动,表明制得的CACCDs主要含C、N、O等元素且碳点表面有较多的-OH和-NH。

图 1 CACCDs表征图
由图2为CACCDs的紫外吸收和荧光光谱图,可以看出,CACCDs紫外吸收光谱的最大紫外波长在280 nm处,由碳点C=C的π~π*跃迁产生。CACCDs的荧光强度随着激发波长的增加而增强,而当激发波长超过410 nm后,荧光强度反而随着激发波长的增加呈明显降低的趋势,这可能是因为激发能量不足所致;以罗丹明6G为参比,计算得CACCDs量子产率约为26.00%。以上分析可知,CACCDs荧光性能良好。

图 2 CACCDs的紫外吸收和和荧光光谱图
2.3 CACCDs的荧光稳定性
实验考察了室温下CACCDs贮存时间、紫外照射时间、体系pH值、盐浓度、常见潜在共存物质(K+、Na+、Ca2+、Mg2+、Cu2+、Fe3+、蔗糖、葡萄糖、甘氨酸、亮氨酸、丙氨酸等)的影响。结果发现,以上条件对CACCDs的荧光强度几乎无影响,表明该碳点具有良好的荧光稳定性。
2.4 DX1002猝灭CACCDs荧光的定量关系
实验条件下 CACCDs于 λex/λem=400/530处与DX1002猝灭效果最佳,此处CACCDs有较强荧光并在一定范围内随DX1002溶液浓度的增加而减弱,如图3所示。因此,后续实验选择激发波长为400 nm,发射波长为530 nm。

图 3 CACCDs中加入不同浓度DX1002的荧光强度变化
2.5 DX1002检测条件优化
2.5.1 缓冲体系的影响
实验考察了BR缓冲溶液体系pH值(2、3、4、5.8、6.8、7.4、7.8、8、9)对 DX1002检测的影响(图4),结果表明,pH的变化对CACCDs与DX1002体系荧光猝灭程度无明显影响,因此,体系中BR缓冲液可视情况加入。

图 4 pH值对DX1002-CACCDs体系荧光强度影响
2.5.2 碳点CACCDs浓度优化
考察CACCDs浓度对体系荧光测定结果的影响 (图 5),当 CACCDs的浓度为 0.250 mg/mL时,直线斜率最大,灵敏度最高。
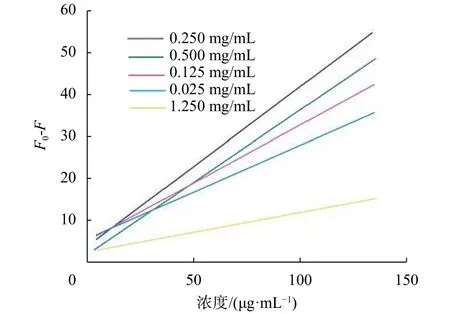
图 5 CACCDs的浓度对DX1002-CACCDs体系的影响
2.5.3 反应时间优化
室温下考察反应时间对体系的影响,发现CACCDs荧光强度在加入DX1002后立即下降,猝灭程度几乎不受反应时间影响。
2.6 方法学验证
2.6.1 专属性
1)常见共存物质影响
当相对误差在±5.0%以内,CACCDs浓度为250 μg/mL,DX1002 浓度为 50 μg/mL 时,考察了常见细胞阳离子、微量金属离子、糖类、氨基酸等潜在共存物质对测定结果的影响,结果见表2。

表 2 共存物质的影响
可看出,上述潜在共存物质对DX1002-CACCDs体系的荧光信号几乎无影响,说明方法选择性及抗干扰能力好,可用来检测DX1002的含量。
2)有关物质的影响
将起始原料异香草醛、三甲氧基苯乙酸以及DX1002顺式异构体代替DX1002分别加入待测体系中,考察其对CACCDs荧光强度的影响。结果表明,异香草醛和DX1002顺式异构体可猝灭CACCDs荧光,且在一定范围内存在线性关系,三甲氧基苯乙酸对CACCDs荧光强度干扰较小。杂质的限度要求为0.1%,上述物质在该含量存在时均不会干扰DX1002的测定,方法专属性良好。
2.6.2 标准曲线、检出限及精密度
在最佳实验条件下绘制标准曲线,结果表明,体系 ΔF(ΔF=F0-F)与DX1002 浓度在 2.5~75 μg/mL范围内呈良好的线性关系,线性回归方程为ΔF=0.805C-0.506 8,相关系数r2=0.998 8,检出限为1.16 μg/mL。对同一供试品溶液进行6次平行测定,方法的相对标准偏差为0.82%。
2.6.3 重复性试验
分别测定不同浓度的DX1002(20181201)供试品溶液,按“1.2.3”项下DX1002测定方法操作,结果表明在24.99~49.98 μg/mL DX1002浓度范围的低、中、高浓度溶液中重复性精密度为1.89%。
2.6.4 加标回收率实验
取重复性中配制的高、中、低浓度DX1002(20181201)供试品溶液各3份,按1.2.3的DX1002测定方法操作,结果如表3所示,低、中、高浓度DX1002溶液的平均回收率为101.7%(RSD=2.08%)。

表 3 回收率结果
2.7 样品测定
利用本法对三批DX1002样品(20181201)、(20180501)、(20180301)进行测定,结果如表 4。

表 4 样品测定结果%
2.8 与HPLC含量测定结果的比较
2.8.1 HPLC测定法
取 DX1002(20181201)原料药细粉 250 mg,精密称定,加适量流动相超声,完全溶解后转移至100 mL的棕色容量瓶中,流动相定容至刻度。精密量取适量,加流动相稀释成每毫升含25 μg DX1002原料药的供试品溶液,备用。另取DX1002对照品250 mg,精密称定,同法,加流动相稀释成每毫升含25 μg DX1002对照品溶液,备用。分别精密量取DX1002的对照品溶液和供试品溶液各20μL,注入液相色谱仪,以 Platisil ODS 柱(150mm×4.6mm,5μm)为色谱柱,0.1%甲酸甲醇∶0.1%甲酸水溶液=70∶30(V/V)为流动相,0.5 mL/min的流量进行分离,于314 nm检测,记录色谱图,以峰面积外标法计算DX1002的含量,测得DX1002(20181201)原料药的平均含量为99.4%(RSD=1.19%)。
2.8.2 方法比较
由表5可知,HPLC法与本法测得的DX1002的含量基本吻合(P>0.05),RSD小于2%,单样本分析时间比较可知,本法分析速度较HPLC法快75倍,体现出本法的高通量、特异性、高灵敏度等特点。

表 5 两种方法含量测定结果1)(n=3)
3 结束语
针对肿瘤血管抑制剂DX1002的含量测定,本文构建了一种基于新型纳米碳点材料(CDs)的特异性荧光定量方法。以柠檬酸、尿素为原料,经工艺优化、条件筛选,得到自发荧光可被DX1002特异性猝灭的CACCDs,且在一定范围内,体系ΔF值(ΔF=F0-F)与DX1002浓度存在良好的线性关系,以此为根据构建了DX1002体外特异性荧光定量方法。本法可实现DX1002含量的快速、灵敏、特异性体外定量检测,检测结果与经典的HPLC法基本吻合(P>0.05),分析速度却较HPLC法快约75倍。体内分析常采用的色谱法或色-质联用等方法存在着生物样本前处理繁琐、分析耗时长、灵敏度低等缺陷,本文利用DX1002与碳量子点间存在的荧光定量猝灭关系建立的体外定量方法,可为DX1002高通量特异性体内分析检测方法的建立提供依据,并为其他与荧光碳点存在定量猝灭关系物质的含量测定提供了思路。
