CRISPR/Cas9基因编辑技术在作物中的应用
2021-04-22林萌萌李春娟闫彩霞孙全喜赵小波苑翠玲单世华
林萌萌 李春娟 闫彩霞 孙全喜 赵小波 王 娟 苑翠玲 单世华
(山东省花生研究所,山东 青岛 266100)
随着基因组测序技术的不断发展,不同生物携带的遗传信息已经触手可及,利用基因编辑技术,人类实现了对生物遗传信息的定向操控。基因编辑技术的发展经历了漫长的过程,由最初的大范围核酸酶、锌指核酸酶(zinc finger nucleases, ZFNs)、类转录激活因子效应物核酸酶(transcription-like activator effector nucleases, TALENs)到CRISPR/Cas9(clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9, Cas9),基因编辑技术的编辑效率和操作性不断提高,引起了科学界的关注[1]。与以往的技术相比,CRISPR/Cas9技术操作更简单,应用潜能巨大,被《科学》杂志评为2013年十大科学突破之一。目前,该技术已被广泛应用于基础科学、人类疾病治疗、作物基因功能及遗传育种等领域的研究中。
基因编辑技术的本质是在基因组特定部位造成DNA双链断裂(double-strand breaks, DSBs),断裂的DNA在修复时容易产生错误,造成碱基的插入或删除,从而影响基因的功能。本文就CRISPR/Cas9基因编辑技术的产生、发展和应用情况进行了系统阐述,旨在为利用该技术进行农作物种质创新、基因挖掘和育种提供指导与帮助。
1 CRISPR/Cas9技术的产生
1.1 基因编辑技术的产生
真核生物的基因组中拥有数量庞大的遗传信息,实现对基因序列的操控对于生物学和医学领域的研究都具有十分重要的意义。20世纪70年代研究人员首次发现细菌利用限制性核酸内切酶系统抵御病毒,这给DNA重组技术提供了思路[2-4],据此科学家实现了生物体外的DNA操控。1983年Rothstein[5]首先对酵母细胞特定基因进行敲除,实现了真核生物活细胞的基因编辑。两年后Capecchi[6]和Smithies等[7]在哺乳动物细胞中通过同源重组的方式实现了外源基因的定点插入。外源基因敲入是研究模式生物基因功能的有效方式,然而基因整合效率低[6]、特异性差[8]等问题影响了其应用。
研究者发现通过在靶位点处引起DNA双链断裂可以大大增加基因整合的特异性[9]。在早期的研究中,研究者利用切割位点少的大范围核酸酶,如识别序列为18 bp的核酸酶Ⅰ-SceⅠ,在老鼠的基因组内引入DNA双链断裂[9]。尽管这种大范围核酸酶增加了基因编辑的效率,但是其识别序列较长,靶向基因内存在识别位点的概率极低。
1987年Klug等[10]发现了锌指蛋白,这为基因靶向编辑开辟了新纪元。锌指结构是锌离子控制的可自我折叠成“手指”形状的多肽空间构型,可以结合在DNA的特定位置,每个锌指结构可以识别3个碱基序列。因此,多个锌指结构可以装配成复合体来实现DNA的特异结合。通过融合锌指蛋白和FokⅠ的DNA切割结构域,研究人员装配出锌指核酸酶[11]。实践证明,锌指核酸酶系统不仅可以在模式生物中大大提高同源重组效率,在人类细胞中也有同样效果[12-13]。随着基因编辑研究的逐渐深入,2009年Boch等[14]和Moscou等[15]发现了来自黄单胞菌属(Xanthomonas)的一种类转录激活因子效应物(transcription activator-like effector, TALE)可以特异性地识别单个碱基。同锌指核酸酶类似,研究人员将TALE模块与FokⅠ的DNA切割结构域融合,产生了类转录激活因子效应物核酸酶[16-18]。
1.2 CRISPR/Cas9技术的发现
尽管大范围核酸酶、锌指核酸酶以及后来的类转录激活因子效应物核酸酶提高了基因编辑的效率,但是在应用上仍存在困难。锌指核酸酶和类转录激活因子效应物核酸酶在靶向不同的位点时,需要重新设计一系列的蛋白来匹配不同的靶点序列,实际操作复杂繁琐,应用门槛较高。而CRISPR/Cas9基因编辑技术以简便的操作和极高的效率在技术上实现了革新(图1)。
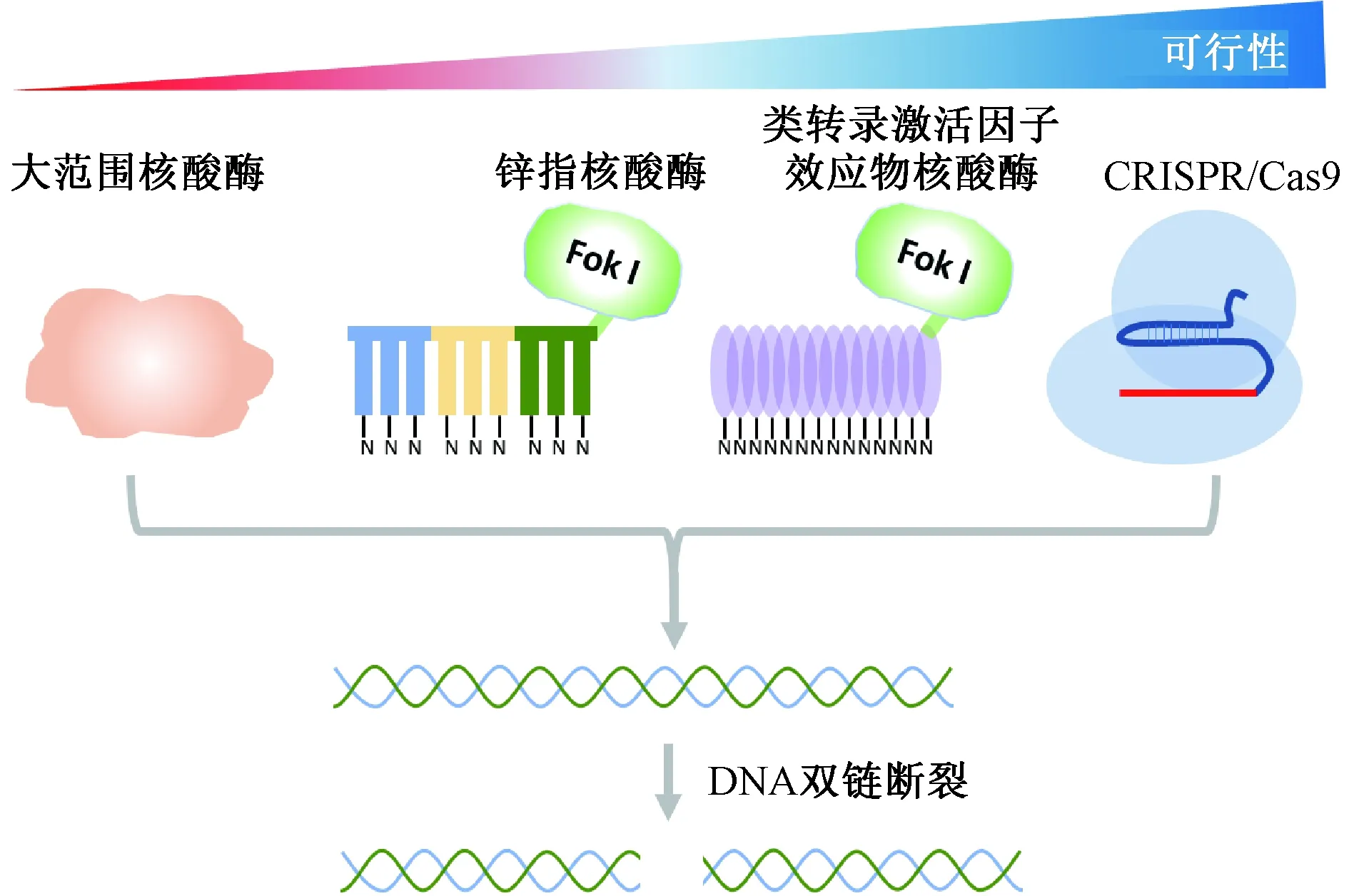
注:大范围核酸酶是经过改造的限制酶,可以识别一段长DNA序列;每个锌指核酸酶单元识别3个DNA碱基;每个TALE单元识别单个碱基;CRISPR根据sgRNA(small guide RNA)和PAM序列的位置决定靶点。这4种技术都可以引起DNA双链断裂,进而引发不同的DNA修复方式。
CRISPR的全称是成簇规律间隔短回文重复序列[20],它是由小导向RNA(small guide, sgRNA)介导的基因组定向编辑技术。1987年,日本科学家首先在大肠杆菌中发现了CRISPR序列[21],其重复序列(repeats)被多个不同的间隔序列(spacers)隔开。计算机分析发现CRISPR序列存在于40%的细菌与90%的古生菌中[22],这些序列临近多个保守的CRISPR关联蛋白(CRISPR-associated,Cas)基因[20],而且间隔序列均来自于病毒或者外源质粒[22-24]。研究者认为该系统与细菌抵抗外来质粒或者噬菌体有关[22-23,25]。2007年,Rodolphe等[26]首先通过试验证明了CRISPR系统的工作机制:经过病毒侵染后,细菌的基因组内整合了一段从病毒基因组中获得的DNA间隔序列,且这段CRISPR位点内的DNA间隔序列可以指引Cas酶抵抗这种病毒。一年后,Brouns等[27]揭示了由间隔序列转录出的crRNA(CRISPR RNAs)可以对Cas酶进行操控。2008年,Deveau等[28]发现不同间隔序列的临近序列非常相似,这段称为PAM(protospacer adjacent motifs)的序列对CRISPR系统的工作非常重要。 Garneau等[29]发现在众多Cas蛋白中,只有Cas9在嗜热链球菌(Streptococcusthermophilus)中有DNA催化活性。这些发现都为CRISPR系统成为一种生物技术工具奠定了基础。2011年,Deltcheva等[30]揭示了Cas9酶的催化受两条短的RNA控制。此后,CRISPR系统被证明可以作为基因编辑的工具靶向细菌中特定的DNA序列[31-32]。Jinek等[31]对CRISPR系统进行了简化,使用一条短的sgRNA来替代原来的crRNA和tracrRNA(trans-activation crRNA)。多项研究证明,CRISPR系统可以用于哺乳动物活细胞的基因编辑,展开了基因编辑的新篇章[33-35]。此后,科学家可以通过设计sgRNA序列来特异靶向不同基因,CRISPR作为一项前所未有的基因编辑技术得到广泛应用。本文统计了2005—2019年关于CRISPR的论文,发现相关CRISPR的研究论文数目逐年增加,合计超过15 700篇(图2)。
在细菌和病毒数亿年的斗争中,细菌进化出了多种CRISPR免疫响应系统。不同CRISPR系统主要依据Cas基因的结构来划分[36-37],目前CRISPR系统主要被分为两大类,每一类又包括多种不同的CRISPR类型。第一类包括Ⅰ型和Ⅲ型CRISPR系统,第二类包括Ⅱ型、Ⅳ型、Ⅴ型和Ⅵ型CRISPR系统[38]。尽管CRISPR/Cas系统类型众多,目前广泛应用类型是来自于酿脓链球菌(Streptococcuspyogenes)的Ⅱ型CRISPR/Cas9系统,该型系统的PAM序列为简单的5′-NGG-3′,应用较方便。
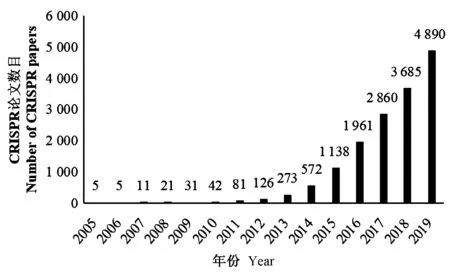
注:2005-2019年PubMed数据库收录的标题或摘要中含有“CRISPR”或“Cas9”的文献数目。
2 CRISPR/Cas9技术基因编辑特点
2.1 DNA断裂的修复方式
CRISPR/Cas9系统包含sgRNA和Cas9核酸酶两部分:sgRNA 5’端1~20 bp的序列为靶点识别序列,通过互补配对决定DNA的切割位置,Cas9核酸酶切割DNA双链[31]。该技术造成的DNA双链断裂(DSBs)主要通过非同源末端连接(nonhomologous end-joining, NHEJ)或同源重组(homology-directed repair, HDR)两种方式进行修复[39-40]。
NHEJ修复途径不依赖DNA同源性,直接将断裂的DNA连接,由此产生的DNA修复错误可能导致基因功能丧失(图3-a)。该修复途径易在DNA断裂处引入或丢失一个至多个碱基,造成基因突变[41]。在提供外源基因作为Donor的情况下,NHEJ修复途径也可以实现基因插入(图3-b)或基因替换(图3-c)[42]。当基因存在两个靶点时,DNA的断裂可能导致两靶点间DNA大片段删除(图3-d)[42]。
HDR修复是以含有同源序列的DNA作为模板进行的精确修复方式[43]。姐妹染色单体、同源染色体,或者经过人工修改后具有同源序列的外源DNA都可以作为HDR修复的模板[43]。因此,研究人员可以通过提供含有同源序列的Donor实现基因替换或基因插入(图3-e)[44]。

注:DNA断裂双链通过NHEJ和HDR两种方式进行修复。NHEJ修复方式可能会造成DNA断裂处的碱基插入或缺失(红色),产生基因突变(a),外源的DNA供体可能被错误连入基因,造成基因插入(b)。两个靶点造成的断裂可能造成基因替换(c)或者大片段 丢失(d)[43]。以同源片段作为模板的HDR修复方式是精确的,可以通过提供Donor实现基因替换或基因插入(e)。
2.2 基因的编辑效率
研究表明,利用CRISPR/Cas9技术对水稻基因进行编辑,不同靶点的编辑效率不同,编辑效率为21%~67%,其中53.9%的基因编辑类型为单碱基插入,插入的碱基绝大多数为A或T[45]。研究人员对水稻不同基因上的46个靶点进行了编辑,克隆测序结果表明,328条测序结果中280条含有不同类型的突变,177条为双等位基因突变,81条为纯合突变,19条为杂合突变,主要的突变方式是A或T单碱基的插入(54.1%),不同靶点的编辑效率与靶点的GC含量息息相关[46]。在Shan等[47]的研究中,转基因水稻中的基因编辑效率为4.0%~9.4%。由此可见,不同靶点的基因编辑效率也是不同的,与靶点序列及其所处位置的染色质状态、靶点序列、外源载体在染色质上的整合位置等有密切关系。
2.3 CRISPR/Cas9技术脱靶问题
在自然界中,细菌的CRISPR系统主要用于抵御病毒的侵染。由于病毒的变异速度较快,所以特异性较低的CRISPR系统可能对细菌更加有利。早期的一些研究证实了这种现象的存在,也说明CRISPR系统作为一种基因编辑工具存在着脱靶的风险[48-52]。 Kuscu等[53]和Wu等[54]通过染色质免疫沉淀和高通量测序(chromatin immunoprecipitation and sequencing, ChIP-seq)的方法对脱靶现象进行了研究,结果表明,Cas9的结合位点主要位于染色体的开放区域,PAM序列远端的碱基容许一定程度的错配,但并不是所有的Cas9蛋白的结合位点都会进行DNA切割,DNA切割对序列的特异性要求更加严格。靶位点序列的特异性决定了脱靶情况发生的可能性,靶点序列与其同源序列在PAM邻近处有大于2 bp的差异,在整个靶序列中有5 bp的差异可有效避免脱靶问题[31, 50]。对于CRISPR/Cas系统来说,PAM序列对于靶位点的识别至关重要,SpCas9的变体SpCas9-NG可将5′-NGG-3′ PAM扩展为5′-NG-3′PAM,有效缓解PAM序列的限制问题[55]。而近期的研究表明,SpCas9-NG可能导致自体靶向编辑,自编辑产生突变的sgRNA仍具有靶向能力,会导致意外的脱靶事件[56]。
基因转化后的CRISPR/Cas9载体可能会整合到基因组中,使sgRNA和Cas9持续表达,增加了脱靶风险[43]。目前,可以通过转化核糖核蛋白(ribonucleoproteins, RNPs)复合体的方法来避免这个问题,同时也避免了转基因的问题[57-59]。通过全基因组测序的方法可以检测脱靶的情况,Zhang等[45]对基因编辑的水稻进行了全基因组测序以检测脱靶问题,结果所有靶点均未出现严重的脱靶问题。除了基因组水平的深度测序,BLESS[60]、GUIDE-Seq[61]和Digenome-Seq[62]等多种测序方式也能有效地找到基因组突变位点,检测基因编辑的情况和脱靶问题。
在植物的基因编辑中,通过设计特异性较高的靶位点可以降低或避免脱靶情况的发生[63],也可以通过回交或杂交方式将非目标突变分离出去。2020年,Manghwar等[64]对基因编辑脱靶现象的机理和存在的问题进行了阐述,总结了脱靶效应的评估方法等,为降低脱靶风险提供了一定的指导。
3 CRISPR/Cas9技术在作物中的应用
3.1 利用CRISPR/Cas9进行基因突变
随着CRISPR/Cas9技术在哺乳动物细胞基因编辑的应用,经过改造的CRISPR/Cas9载体也很快用于拟南芥[65-67]、烟草[68-69]、水稻[70-72]、小麦[73]和玉米[74]等植物基因组的定向编辑研究。
Mao等[75]首先对CRISPR/Cas9表达盒进行优化,构建了可以在拟南芥和水稻中表达的植物适应性表达盒,并成功对拟南芥和水稻中的基因进行了编辑。2013年,Feng等[76]再次使用由CaMV 35S启动子引导的Cas9和AtU6-26启动子或OsU6-2启动子连接的sgRNA对拟南芥和水稻进行基因编辑,产生了稳定的转基因编辑植株,9个靶位点的基因编辑效率介于5%~84%之间。2014年,Feng等[77]对拟南芥的7个基因进行编辑,并且对基因编辑植株的遗传情况进行研究,发现T1、T2和T3植物携带突变的比例分别为71.2%、58.3%和79.4%,T1的突变杂合体在T2产生了22%的突变纯合体,所有纯合突变均能稳定地传递至下一代。
CRISPR/Cas9载体和转化方式的优化也在逐步进行。Miao等[71]对CRISPR/Cas9系统进行了优化,基因的靶点序列首先被克隆到sgRNA的表达载体中,然后再克隆到含有Cas9基因的终载体中。研究人员对水稻叶绿素合成基因CAO1和分蘖夹角基因LAZY1进行定点突变,基因编辑效率分别为83.3%和91.6%。2015年Ma等[46]对Cas9基因的密码子进行优化,并采用Golden Gate Cloning方法构建了适用于单子叶植物和双子叶植物的高效CRISPR/Cas9载体,成功对水稻和拟南芥的基因进行编辑,大多数突变为双等位基因突变和纯合突变。Xie等[78]将tRNA和gRNA嵌合到一起,开发了从一个多顺反子基因生产大量sgRNA的通用策略,成功实现同时对水稻基因组中多个位点的编辑。Gil-Humanes等[79]使用小麦矮病毒复制子作为CRISPR转化工具,使小麦基因靶向效率提高了10倍以上。
CRISPR/Cas9技术的转基因安全问题和脱靶问题一直是人们关注重点。2016年,Zhang等[80]通过小麦中瞬时表达CRISPR/Cas9的DNA或RNA,有效地减少了外源基因的整合。2017年,Liang等[57]另辟蹊径,通过直接向小麦幼胚轰击CRISPR/Cas9的核糖核蛋白(RNPs)复合体的方法来避免转基因的产生,结果发现该方式可以对靶基因进行编辑并且有效减少了脱靶效应。
在花生突变体研究中,通常使用化学诱变的方式诱导产生突变体[81]。2019年,Yuan等[82]率先通过CRISPR/Cas9技术对花生的脂肪酸去饱和酶ahFAD2基因进行了突变,该研究利用花生的原生质体和发根作为转化材料,实现了对花生的基因编辑。
3.2 利用CRISPR/Cas9进行精确基因编辑
利用CRISPR/Cas9技术对基因进行突变操作简便,然而实现精确的单碱基编辑、基因替换或者基因插入则相对复杂。为此,科学家尝试了不同的策略,早期主要通过提供外源的DNA作为修复模板或者基因替换的模板进行精确编辑,后期逐渐形成的第二代CRISPR/Cas9基因编辑技术更容易实现单碱基编辑。2013年,Li等[44]将烟草NbPDS基因编辑载体和双链DNA Donor共转化,利用Donor作为修复模板,通过同源重组途径实现基因替换。为了防止DNA断裂处的修复错误,2016年,Li等[42]在内含子区域选择靶位点,转化的同时提供基因编辑载体和外源DNA Donor,通过非同源末端连接途径实现了水稻OsEPSPS基因的替换,获得了抗除草剂的植株;另一方面,也通过提供Donor实现了1.6 kb的基因插入。2020年,Dong等[83]通过CRISPR/Cas9技术实现了在水稻基因组中的安全位置插入5.2 kb类胡萝卜素生物合成元件。研究表明,该水稻种子中的类胡萝卜素含量高,并且形态及产量特性与野生型相似,为作物精确的基因敲入提供了思路[83]。除了DNA双链或者环形质粒可以作为Donor以外,单链DNA片段也可以作为DNA修复模板[47]。
野生型的Cas9酶可以造成DNA双链断裂,而第二代基因编辑工具以Cas9切口酶为基础,可以在不产生DNA双链断裂的情况下实现靶位点的碱基替换,使单碱基编辑更易实现。这种单碱基编辑工具可以实现胞嘧啶(cytosine, C)到胸腺嘧啶(thymine, T)和腺嘌呤(adenine, A)到鸟嘌呤(guanine, G)的碱基替换[84-86]。2016年,Komor等[86]发现将Cas9切口酶与APOBEC1脱氨酶和尿嘧啶糖基化酶抑制剂(uricall glycosylase inhibitor, UGI)蛋白融合,可以有效地在靶位点处将C转化为T。2018年,Gaudelli等[84]将RNA腺苷酸脱氨酶融合Cas9切口酶,可以实现靶点碱基A到G的转换。这些新型的单碱基编辑策略极大地扩展了基因编辑技术的应用范围。Kuscu等[87]和Billon等[88]实现了将氨基酸编码的密码子转换为终止密码子,可以提前终止基因的翻译。此外,将活性诱导的腺嘌呤脱氨酶与dCas9(dead Cas9)融合成dCas9-AID复合体,由其引发的基因突变可以作为基因功能筛选的重要手段[89-91]。
3.3 基因突变的检测方式
CRISPR/Cas9基因编辑植株的鉴定是基因编辑过程的重要步骤[43]。目前,基于PCR和酶切的检测方法主要有两种。第一种方式是基于PCR扩增及限制性酶切的PCR/RE(restriction enzyme),Cas9酶切位点(5′-NGG-3′上游3 bp)含限制性酶识别序列时可以使用该方法。完成基因编辑后,通过PCR扩增和限制性酶切反应可以检测靶点处是否发生突变[42-43,92-93]。第二种方式是T7EI(T7 endonuclease Ⅰ)酶切检测,此方法不需要考虑靶位点是否含有限制性酶识别序列[92,94]。以上两种方法虽能检测出突变植株,但是要了解基因编辑情况还需通过测序。利用叠峰分析软件,可以分析突变植株的PCR产物测序结果[95]。
通过PCR和酶切方式检测突变体相对耗时耗力,Peng等[96]开发了基于实时荧光定量PCR(quantitative real-time PCR, qPCR)的检测方式,无需酶切就能从样品中找出突变体。利用该方法,研究人员成功将水稻、拟南芥、高粱和玉米的突变体从野生型中分离出来。随着新一代测序技术的发展,高通量测序的费用已经大幅度降低,准确性提高[43]。通过第二代测序的方法可以大批量检测CRISPR/Cas9引起的突变,是一个简便、高效的选择[97-98]。
4 CRISPR技术的其他应用
4.1 基因表达调控
研究发现,没有切割活性的dCas9仍可以紧密结合DNA,这种紧密的结合可干扰转录因子和RNA聚合酶Ⅱ与DNA结合[99],由此开发的CRISPR干扰技术可以干扰转录过程,从而抑制基因表达[99]。在此基础上,将dCas9与有转录调控功能的转录激活因子或转录抑制因子融合,可以有效激活或抑制基因表达[100-101]。
4.2 表观遗传学、基因座定位、染色体结构研究
在表观基因组学研究中,将dCas9与DNA甲基转移酶融合可以调控特定基因区域的甲基化情况,进行表观遗传学研究[102-105]。
荧光杂交技术(fluorescent in-situ hybridization, FISH)是常用的基因座定位方式[106-107],但由于该技术需要对细胞固定加热,所以很难在活细胞中应用。通过将dCas9连接荧光标签,可以在活细胞中实现特定基因座的定位[108]。
在染色质拓朴结构的研究中,CRISPR用于指引染色质形成环状结构,为探索染色质结构与功能的关系及染色质结构对基因表达调控的影响提供思路[109]。
4.3 基因功能筛选
除了上述应用之外,CRISPR技术还应用于大规模基因功能筛选。该策略需建立包含数千种sgRNA的Cas9/sgRNA池,不同sgRNA靶向基因组的不同位点。通过降低丰度使每个细胞只接收到一种sgRNA。高通量测序可以筛选出突变基因,将突变的基因与表型关联,获得基因功能的信息[110]。
5 结论与展望
随着CRISPR/Cas9技术的发展,其应用层面也越来越广泛,在作物遗传育种的研究中有广阔的应用前景。该技术操作简便,效率较高,为作物种质创新、基因功能研究提供了有效工具。在多倍体作物的基因编辑中,CRISPR/Cas9技术可以同时敲除多个同源基因,极大地提高了基因突变的特异性和有效性[93],为多倍体作物基因编辑提供了技术手段。在作物种质创新上,目前通过精确的基因编辑已经创造出了抗除草剂水稻[111]和高类胡萝卜素含量的水稻[83]。在未来的研究中,利用CRISPR技术进行精确编辑,研究有价值的生物性状,提高作物中功能成分含量将是一个新的研究方向。
目前,人们对CRISPR/Cas9基因编辑技术的原理、操作过程和基因编辑特点都有了基本的了解,但是对于基因编辑过程中的编辑效率、脱靶现象和遗传性问题仍需要更加深地的研究。此外,虽然CRISPR/Cas9技术应用广泛,但是该技术的转基因安全性问题和脱靶问题仍存在争议。该技术目前已实现在不引入外源基因的情况下进行基因编辑[57],检测不到转基因痕迹,但是如何进行监管仍是政府需要面对的一个问题。将CRISPR/Cas9技术应用于临床治疗不仅存在安全隐患[112],还存在着一系列的伦理争议,这是我们在发展技术的同时需要深思的问题。
