全基因组关联分析在蔬菜育种研究中的应用
2021-04-21曾美娟刘建汀卓玲玲陈敏氡叶新如朱海生温庆放
曾美娟 刘建汀 卓玲玲 陈敏氡 叶新如 王 彬 朱海生 温庆放
(福建省蔬菜遗传育种重点实验室,福建省农业科学院作物研究所,福建省蔬菜工程技术研究中心,福建福州 350013)
随着20 世纪80 年代初基于DNA 的分子标记和80 年代末先进的统计工具的出现,挖掘控制数量性状的基因组区域成为可能。挖掘控制数量性状基因组的方法包括数量性状座位(quantitative trait locus,QTL)区间定位和全基因组关联分析(genome-wide association study,GWAS)。传统的QTL 定位高度依赖于双亲的遗传多样性,检测到QTL 效率因群体而异。QTL 区域也可能相当大,包含太多的基因,很难作为潜在的候选基因进行研究。此外,QTL 定位往往需要构建作图群体,耗时较长且定位精度不高(Rafalski,2010)。GWAS可以在一定程度上克服QTL 分析的局限性,它可以利用自然群体缩小候选区域,同时对多个性状进行分析(Yu &Buckler,2006;Huang &Han,2014),以降低分析误差(曹英杰 等,2019),极大地提高育种效率。全基因组关联分析高效地将表型和基因型进行关联并用于遗传作图和搜寻相关性状候选基因(Gajardo et al.,2015;阙青敏 等,2019),可同时对多个复杂性状进行关联,检测多个等位基因,适用于定位性状关联区间,功能基因研究,开发性状选育标记等,具有高分辨率和高通量等优点,在蔬菜育种研究中的应用日益广泛。全基因组关联分析在揭示蔬菜复杂性状的分子机理和蔬菜分子育种中起到重要的作用(李廷雨 等,2020)。
1 全基因组关联分析
1.1 概述
全基因组关联分析是近年来兴起的遗传分析方法,其以连锁不平衡(linkage disequilibrium,LD)为基础,通过识别数百个或数千个个体定位群体中高密度的分子标记,一般是上万个甚至上百万个单核苷酸多态性(single nucleotide polymorphism,SNP)标记,筛选出与复杂性状表现型变异相关联的分子标记(Du et al.,2018)。换言之,通过关联分析分子标记与性状变异,对群体中的个体进行大规模的基因分型和表型分析,从而识别导致个体之间表型差异的基因组区域(Hirschhorn &Daly,2005;Huang et al.,2010;Zhao et al.,2011)。连锁不平衡是指群体内不同位点上等位基因间的非随机关联(曹英杰 等,2019)。连锁不平衡是GWAS分析的基础(Abecasis &Cookson,2000),受多因素的影响,不同物种间基因组中连锁不平衡也存在显著差异。高LD 水平的群体,能够缩减GWAS分析时所需的群体数量。近年来,随着以SNP 为代表的第3 代分子标记技术的发展,大大加快了GWAS 的发展速度,几千甚至几百万个标记被用于1 个GWAS,在人类和动植物复杂性状遗传研究中已取得初步成果(段忠取和朱军,2015),亦已成功应用于多种作物的重要农艺性状的遗传研究(Elshire et al.,2011;赵振卿 等,2014;He et al.,2014)。
1.2 分析策略和常用软件
GWAS 应用于植物育种的研究策略主要包括以下几方面:①选择植物群体材料。群体规模太大,性状调查费用和基因型检测费用均会相应增多。选择表型齐全和遗传变异丰富的研究群体可以减少所需群体内的个体数目,提高关联分析的分辨率。②表型鉴定。设计合理的田间试验,多年多点种植试验材料和多区域重复随机调查表型性状,并将调查结果进行整合(刘坤 等,2018)。③基因型的测定。获取选定试验材料的全基因组SNP 位点,从而实现基因型分析(Seki et al.,2005;Rutkoski et al.,2013;韩德鹏 等,2018)。④ 关联分析。对选定试验群体进行群体结构分析,选用合适的统计分析模型对基因型与表型进行分析(Raman et al.,2019;孙程明 等,2020)。涉及质量性状关联分析时,通常可以采用Logistic 回归模型进行分析;涉及数量性状时,通常可采用普通线性回归模型(卜李那和赵毅强,2019),但数量性状通常受到多种因素的共同影响,在研究过程中通常采用不同的混合模型(表1)。⑤ 候选基因筛选(姜洪真 等,2018)。
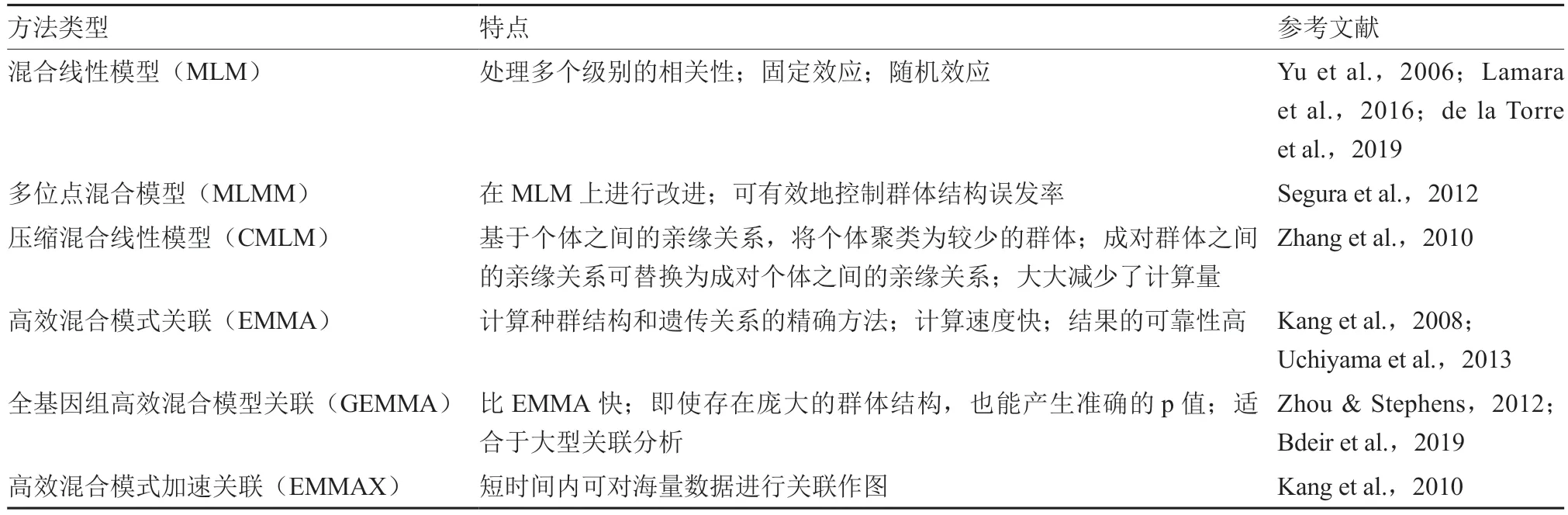
表1 近年来部分GWAS 中采用的混合模型方法及其特点
目前分析过程涉及的软件很多,其中PLINK软件(Purcell et al.,2007)是较早使用的关联分析软件,它可用于复杂数量性状、关联作图、数据转化与处理、LD 分析、单倍型检验等。采用SAMTOOLS 软件(Li et al.,2009)检测、过滤SNP,采用ANNOVAR 软件(Wang et al.,2010)对检测到的SNP 进行注释,采用GCTA 软件(Yang et al.,2011)对群体进行主成分分析、单性状和两相关性状关联分析,采用STRUCTURE 软件(Evanno et al.,2005;王艳玲 等,2017;Volante et al.,2017)对群体结构进行分析。采用GEMMA 软件(Zhou &Stephens,2012)基于SNP 进行关联分析。采用TASSEL 软件(Bradbury et al.,2007)进行各种模型的关联分析,估算LD 值和作图、估测群体结构和绘制基于遗传距离的树状图等。GAPIT 软件(Lipka et al.,2012)用于BLUP 基因组预测关联分析和BLUP 基因组预测。
2 全基因组关联分析在蔬菜育种研究中的应用
2.1 蔬菜生长发育过程相关性状应用研究
前人在蔬菜生长发育过程相关性状方面,如黄瓜发芽期、黄瓜幼苗阶段、白菜类作物抽薹开花、菠菜雌雄同株、普通菜豆相关性状的控制基因等方面都开展了全基因组关联分析。张松等(2019)对黄瓜发芽期进行全基因组关联分析,检测到5 个与相对发芽率关联的位点,2 个与相对发芽势关联的位点,2 个与相对发芽指数关联的位点,2 个与相对胚根长度关联的位点。蔡和序等(2020)对黄瓜幼苗下胚轴长度进行全基因组关联分析,通过分析关联SNP 位点的LD 区间序列,获得Csa1G074930、Csa1G475980、Csa2G381650、Csa3G141820、Csa4G051570、Csa3G627150、Csa5G174640、Csa6G362970等8 个与黄瓜下胚轴长度有关的候选基因,其中既有光形态建成、泛素化、激素信号通路等调控基因,也有调控网络下游参与细胞生长发育,调节细胞大小,直接调控黄瓜下胚轴长度的基因。抽薹开花是白菜类作物关键的农艺性状。白菜类作物主要包括大白菜、普通白菜、芜菁等。挖掘白菜类作物抽薹开花调控位点和基因,对白菜类作物的培育具有重要的意义。龚振平(2016)对182份大白菜自然群体晚抽薹性状进行全基因组关联分析,获得5 个与耐抽薹性状显著关联的位点,为进一步发掘相关性状的候选基因提供了依据。高宝祯等(2017)通过全基因组关联分析鉴定出33 个与白菜类作物开花时间相关的显著关联信号。通过定位出的开花时间候选位点,再根据白菜类作物与同源物种拟南芥的基因共线性关系以及基因功能注释结果初步鉴定出与14 个白菜类作物开花时间相关的候选基因。汪豪英等(2019)通过对82 份菠菜高代自交系的全基因组关联分析,采用压缩混合线性模型在菠菜4 号染色体上检测到1 个强关联区域,并将控制菠菜雌雄同株的基因Xm定位在64.6 kb 的区间内。该范围内存在3 个基因:Spo24600、Spo24601和Spo24602。最近,Wu 等(2020)对来自19 个国家的683 份普通菜豆资源的全基因组进行重测序,发掘出超过480 万个SNP,构建出国际首张精细的普通菜豆单倍型图谱,鉴定出505 个与主要农艺性状紧密相关的遗传位点。
上述研究表明采用全基因组关联分析能够有效鉴定蔬菜生长发育过程相关农艺性状的关键遗传位点。因为蔬菜作物的农艺性状往往都是由多个基因控制且受环境影响,与单基因控制的性状相比,其遗传基础更为复杂。而全基因组关联分析是经典的定量遗传理论的拓展,采用全基因组关联分析的方法能够既简单且快速地鉴定出蔬菜作物控制发育过程相关性状的重要基因,对蔬菜作物农艺性状相关基因的研究以及辅助育种具有重大意义。
2.2 蔬菜品质和产量性状应用研究
在蔬菜品质和产量性状方面,如番茄的果实质量、果实硬度、心室数目、果形指数、代谢物含量以及辣椒的辣椒素含量等相关性状的控制基因都开展了全基因组关联分析。Xu 等(2013)利用关联分析的方法对44 份栽培种番茄、127 份樱桃番茄和17 份醋栗番茄的果实质量、果实硬度、心室数目等性状进行了初步研究,共检测到40 个位点,其中果实质量、心室数目、可溶性固形物等性状的主效位点信号区域都有相关的功能基因。祝光涛(2015)利用843 316 个位点对253 个番茄栽培种亚群中的果实颜色、果实质量和果形指数等5 个性状进行关联分析,发现了11 个明显的关联信号位点,其中1 个果皮颜色位点、2 个果形指数位点和2 个心室数目位点和前人的研究结果一致,另外6个位点为新发现的位点。Bauchet 等(2017)借助10 000 个SNP 标记对300 份番茄的60 种初级和次级代谢产物开展了全基因组关联分析,确定了79个与13 个初级代谢产物和19 个次级代谢产物高度关联的位点。同时,还发现了4 个基因组区域可控制几种代谢物变异,并发掘了决定代谢物含量的候选基因,揭示了番茄亚种复杂而独特的代谢物调控机理。赵建涛(2016)利用混合线性模型对番茄果实中主要的17 种糖酸组分进行全基因组关联分析,共检测到139 个显著关联位点,除了苏糖醇外,在其他16 个糖酸物质上至少检测到1 个显著关联位点。Sauvage 等(2014)利用多位点混合模型对163 份番茄种质进行关联分析,利用遍布全基因组的5 995 个SNP 位点对影响番茄品质的76 种代谢物质进行了分析,检测到了控制19 个性状的44 个显著位点。Nimmakayala 等(2016)使用7 331 个SNP 标记对辣椒性状进行研究,发现72 个SNP 标记与辣椒素含量相关,包括1 个候选基因,该基因编码一种具有与CS 相似的酰基转移酶功能的锚蛋白样蛋白。Han 等(2018)通过QTL 定位和GWAS 挖掘控制辣椒中辣椒素含量的候选基因,共检测到69 个QTL 区域,其中10 个区域与2 个双亲群体的QTL 位于同一位置。在这些区域中,鉴定出5 个已知参与辣椒素生物合成的候选基因。
随着人们生活水平逐步提高,多样化的蔬菜品种给予了大众更多的选择,大众对蔬菜品质的要求则越来越高。在蔬菜育种中,品质高低影响着该品种能否适应市场以及满足消费者的需求。蔬菜品质育种也是蔬菜遗传改良的重点。同时,蔬菜产量的高低也影响着其经济价值,通过全基因组关联分析来开展蔬菜品质和产量性状的相关研究对蔬菜品质和产量的提升具有重要意义。
2.3 蔬菜抗性性状应用研究
为有效控制蔬菜病害的发生与危害,选育和利用具有相关抗性的蔬菜品种也是育种的关键。随着全基因组学的发展,全基因组关联分析也在马铃薯抗晚疫病、大白菜抗霜霉病、瓠瓜抗白粉病、黄瓜抗低温等蔬菜抗性相关基因的挖掘中得到应用。为了明确马铃薯晚疫病抗性的稳定性与标记间的关联,Lindgvist-Kreuze 等(2014)结合田间晚疫病表型数据,对适应热带高地的马铃薯群体的基因型进行全基因组关联分析,发现第9 号染色体上的SNP标记与马铃薯晚疫病抗性相关,且与稳定性有关。龚振平(2016)对182 份自交系材料组成的大白菜自然群体开展5 种病害的全基因组关联分析,分别获得与霜霉病(2 个)、病毒病(5 个)、黑腐病(2 个)、黄萎病(5 个)和根肿病(8 个)抗性显著关联的22 个位点或热点区。吴晓花等(2020)利用两年的抗病表型数据,对117 份瓠瓜微核心种质的白粉病抗性进行研究,通过全基因组关联分析,分别获得22 个和13 个与白粉病抗性相关的SNP 标记。王伟平等(2019)以黄瓜核心种质为材料开展苗期耐低温鉴定和筛选,并进行全基因组关联分析,挖掘耐低温相关位点。在1、3、4、5 号染色体上分别检测到苗期耐低温位点gLTS1.1、gLTS3.1、gLTS4.1和gLTS5.1。魏爽等(2019)进行了黄瓜苗期耐热性筛选,通过全基因组关联分析共检测到7 个与苗期耐热性相关位点gHII4.1、gHII5.1、gHII5.2、gHII6.1、gHII7.1、gHII4.2、gHII6.2。
施用化学药剂能够在一定程度上控制相应的病虫危害,但也带来环境污染等问题,同时增加农民种植成本,产生蔬菜安全问题。通过全基因组关联分析,将这些性状关联标记用于蔬菜分子辅助育种,有助于选育出具有抗性的蔬菜品种。而选育具有抗性的蔬菜品种又是目前防治相应病害、适应不佳种植环境及选育反季蔬菜较为经济、有效的方法。
3 展望
传统的QTL 定位高度依赖于双亲的遗传多样性,检测到的QTL 效应因群体而异。QTL 区域也可能相当大,包含太多的基因,而利用全基因组关联分析(GWAS)可以利用自然群体缩小候选区域,在一定程度上克服QTL 分析的局限性。当然,GWAS 的应用也存在一定的局限性,例如在自花授粉作物中,用GWAS 定位性状相关基因难以达到单基因水平,一些群体结构因素易导致假阳性,非遗传因素产生的表型变化导致的假阳性以及基因与环境的互作效应亦会影响GWAS 结果(Stacey &Joanna,2013)。为获得更精准的GWAS 结果,研究人员需对群体结构加以准确分析,通过增大群体规模来尽可能减少假阳性。相关技术的不断更新也将在一定程度上降低群体结构的干扰。同时,也有必要对结果进行验证(Zhu et al.,2008;Korte &Farlow,2013)。由于每种方法都有其局限性,将不同方法进行联合,取长补短可在一定程度上提高分析结果的准确性。已有研究表明,QTL 定位联合GWAS 是鉴定控制复杂性状的基因位点的一种强有力的组合方法(Han et al.,2018)。
GWAS 只识别与目标性状相关的基因组区域,而不是发现基因,一个性状相关的基因组区域内会有许多标记(例如,多个SNP 标记)。随着下一代测序技术的发展,几千甚至几百万个标记被用于1个GWAS,每个GWAS 可用的标记数量大幅增加,SNP 集被应用于GWAS 是当前的关键研究领域,也将促进GWAS 的发展。近年来,GWAS 研究取得了重大进展,这些性状关联标记也被有效地用于标记辅助选择,以补充传统的蔬菜育种方法,改良简单和复杂的数量性状。GWAS 将极大推动蔬菜由传统育种向高效、定向的分子设计育种转变,亦将为揭示蔬菜主要农艺性状、品质性状和抗性性状的分子机理发挥重要的作用。
