燃煤烟气中SO3与NH4HSO4生成特性及其控制方法研究进展
2021-04-20尹子骏苏胜王中辉王乐乐安晓雪赵志刚陈逸峰刘涛汪一胡松向军
尹子骏,苏胜,王中辉,王乐乐,安晓雪,赵志刚,陈逸峰,刘涛,汪一,胡松,向军
(1 华中科技大学煤燃烧国家重点实验室,湖北武汉430074;2 西安热工研究院有限公司苏州分公司,江苏苏州215153)
近年来,燃煤烟气选择性催化还原(SCR)脱硝技术广泛应用,SCR 脱硝催化剂在将烟气中NO还原脱除的同时,也会导致烟气中一部分SO2催化氧化生成SO3[1]。SO3的生成对燃煤电厂的运行具有显著的不利影响。SO3会与SCR 系统逃逸的NH3结合形成硫酸氢铵(ABS),这是一种黏性很强的物质,沉积在催化剂表面使催化剂中毒,同时也会黏附在下游空预器上,造成其堵塞[2]。当烟气温度降至酸露点以下时,SO3开始冷凝,从而会对下游管道系统和设备造成严重腐蚀[3]。
另一方面,SO3排放也会造成严重的环境问题。烟气中的SO3极易与水分子结合形成H2SO4蒸气。随着烟气温度的降低,H2SO4蒸气会凝结成亚微米级别的气溶胶酸雾。由于硫酸气溶胶的粒径细小,因此难以通过脱硫装置洗涤有效脱除,脱硫系统中总体SO3脱除效率仅为30%~50%[4];而且当烟气中H2SO4质量分数达1.0×10-5~2.0×10-5时就会出现蓝羽现象[5]。SO3排放到大气中会形成酸雨,会腐蚀建筑以及农作物,严重危害人们的身体健康[6]。
尽管目前对SO3的排放标准还没有达成共识,但对SO3的排放控制已受到很多国家重视,并制定了控制SO3排放的相应标准。美国有23个州制定了燃煤电厂SO3排放标准,其中最严格排放限值为0.6mg/m3。德国现有发电厂(机组容量>300MW)的日均SOx(SO2+SO3)排放限值为200mg/m3。在我国,上海在2016年公布了首个SO3排放标准,设定了5mg/m3的排放限值。北京市在2017年公布了SO3排放标准,排放限值为5mg/m3。杭州在2018 年公布了SO3排放标准,排放限值也为5mg/m3[7]。
近些年来,国内外学者针对燃煤烟气中SO3和硫酸氢铵的生成机理与控制方法也进行了一些研究。本文从燃煤烟气中SO3生成特性与转化规律、硫酸氢铵(ABS)生成机理以及SO3与ABS 生成控制方法等三个方面综述分析了相关研究进展,希望为燃煤烟气中SO3与ABS 生成控制深入研究提供基础。
1 燃煤烟气中SO3的生成机理
在燃烧过程中,煤中的大部分硫元素被氧化成SO2,只有较少部分硫元素会在锅炉中进一步氧化成SO3;同时,生成的一部分SO2在经过SCR 脱硝系统时会被SCR催化剂进一步氧化为SO3[8]。
1.1 炉膛内SO3的生成
1.1.1 燃烧过程中SO3均相生成
在煤燃烧过程中,SO3可由均匀气相反应以及非均相反应生成。其中通过均相反应生成的SO3占炉膛内SO3生成总量60%左右,而通过非均相反应生成的SO3比例相对较小[9]。煤燃烧过程中,烟气中过量氧气通过一些反应能在火焰中形成O 自由基,这些O自由基能使SO2氧化成SO3。Hindiyarti等[10]确定了燃烧中气相SO3生成一些关键反应如式(1)~式(4)所示。反应过程中SO2与O 自由基结合,通过与第三体(M)的相互作用形成SO3,SO3形成后可与H等自由基反应产生分解;同时SO2也可与OH 自由基反应生成HOSO2,并进一步氧化生成SO3。
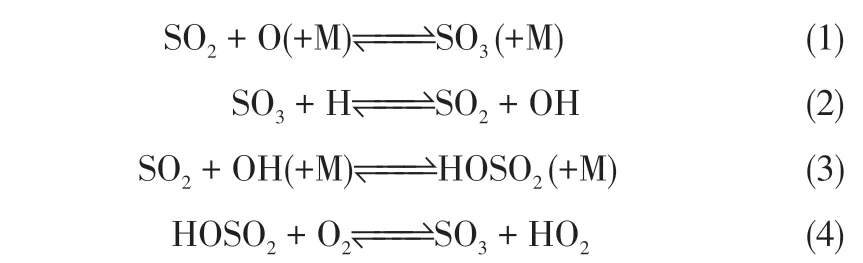
SO3均相反应生成的浓度取决于SO2浓度、O2浓度以及活性基团等[11-12]。SO3的形成与SO2的浓度有着密切的关系,而SO2的浓度则取决于燃料的硫含量和燃烧条件[11]。煤种中含硫量高会导致SO2浓度高,因此烟气中SO3浓度也就越高。如图1 所示[12],随着反应过程内SO2浓度的增加,SO3浓度呈上升趋势,但SO2/SO3转化率随着烟气SO2浓度的增加而降低。较高的O2浓度有利于SO3形成,并且SO3形成速率随着O2含量增加而增加,当达到一个极限值后,会呈现缓慢降低趋势。

图1 O2气氛下SO2浓度对SO3生成的影响[12,22]
1.1.2 燃烧SO3非均相生成
非均相反应生成SO3与均匀气相反应生成SO3同样重要。这种氧化机理取决于SO2浓度、过量空气系数、灰分含量和成分等[13]。飞灰中存在的Fe2O3、Al2O3、CuO 和V2O5等几种金属氧化物对氧化SO2具有催化作用,其中Fe2O3可能是燃煤锅炉产生SO3的主要催化剂[14]。飞灰中Fe2O3氧化SO2与温度的关系很大,如图2 所示,飞灰中SO2/SO3的转化率最大时温度约为700℃,而转化率最低时温度约为400℃。SO3也可以通过与碱土金属氧化物(如CaO和MgO)和碱金属氧化物(如Na2O和K2O)的反应被吸附,从而降低烟气中SO3的浓度[15]。

图2 不同温度下催化剂SO2氧化率[14,22]
1.2 SCR脱硝反应过程中SO3的生成
燃煤电厂除了燃烧过程中会生成一部分SO3,当烟气通过SCR脱硝系统后会有0.5%~1.5%的SO2被催化氧化为SO3[16]。虽然SO2被催化氧化的比例并不高,但由于锅炉炉膛出口SO2尚未经过脱除,其浓度相对较高,因此经过SCR 脱硝系统生成的SO3已经是烟气中SO3主要来源之一。
V2O5-WO3/TiO2催化剂由于其高脱硝活性以及稳定性广泛应用于燃煤电厂SCR脱硝装置中。V2O5作为催化剂的主要活性成分,对NOx脱除起重要作用。同时,研究表明V2O5对烟气中SO2的催化氧化也起到了重要作用。SCR 催化剂中V2O5含量越高,其对SO2氧化率也相应提高[17]。Kamata 等[18]采用红外光谱研究了V2O5/TiO2催化剂表面负载的V2O5对SO2氧化活性,发现与钒原子结合的V=O和V—OH等基团都可参与SO2吸附和氧化反应过程。Ji 等[19]研究了V2O5/TiO2催化剂表面性质与SO2氧化的关系,结果表明,V和Ti离子之间的相互作用改变了V离子的还原过程,在TiO2上负载的V物种更容易被还原。V—OH 和SO2之间的相互作用较弱,而V==O 和Ti—OH 位在SO2的吸附和氧化中起着重要作用。该研究结果提出了可能的反应机理:SO2首先在V==O、V—OH、Ti—OH 活性位上产生弱吸附,形成表面配位的SO32-/HSO3-和SO42-;此外,吸附的SO2被V==O位氧化为SO3,这表明V=O位的末端O2-被转移到SO2;气相中的O2可以取代被消耗的O2-,以便氧化反应可以持续进行;随后,一部分SO3会与Ti—OH反应生成三齿硫酸盐(TiO)3SO==。
Dunn等[20]也提出了SO2在V2O5/TiO2催化剂上的氧化机理,如图3 所示。SO2吸附在V2O5上,并与其上(Ti—O)3V+5==O 物种中的V—O—Ti 配位形成中间体(V+5)·SO2-ads。然后中间体上的V+5—O—SO2键断裂形成气态SO3,最后还原的(V+3)然后被解离吸附的O再次氧化成(V+5)。SO3和SO2在活性中心存在竞争吸附,SO3的优先吸附导致SO3与表面钒的结合力增强。

图3 固体钒催化剂上二氧化硫氧化机理[20]
Xiong 等[21]通过研究H2O、NOx和NH3对SO3生成过程的影响,提出了SO3在复杂气氛中的生成机理。如图4 所示,当SO2被氧化时,V2O5被还原成低价钒(V3+),然后V3+被O2氧化成V5+,从而完成催化循环。气态SO3的形成过程,伴随着高价钒和低价钒的转化,大致可分为三个步骤:①气态SO2通过羟基化学吸附在载体表面;②化学吸附的SO2被高价钒氧化形成吸附的SO3;③吸附的SO3被解吸生成气态SO3。NOx促进了低价钒向高价钒的转化,从而促进了SO2的氧化,使SO3显著增加。反应气氛中NOx的存在对SO2的吸附和SO3的解吸没有明显的影响,而H2O 和NH3的存在则促进了SO2的吸附,但对SO2的氧化没有显著影响。同时,H2O和NH3会分别与三齿硫酸盐结合形成较稳定的二齿硫酸盐和硫酸氢铵,从而抑制了SO3的解吸;NH3对SO3解吸的抑制作用比H2O 明显,对气态SO3的生成有明显抑制作用。

图4 不同气氛下SCR过程中SO3的生成机理[21]
2 烟气中SO3的转化及ABS生成机理
2.1 烟气中SO3的转化特性
SO3在烟气中极易发生反应,图5 显示了燃煤烟气中SO3的迁移与转化路径[22]。炉膛内形成的SO3一部分与H2O 反应转变为气相的H2SO4,这些气相H2SO4在SCR 过程中与NH3发生反应,形成硫酸铵或硫酸氢铵,导致SCR 催化剂失活和空气预热器堵塞。当烟气的温度低于硫酸的酸露点时,另一部分没有反应的气相H2SO4会冷凝形成硫酸液滴,从而对空预器等下游设备造成低温腐蚀。
表1 为杨用龙等[23]测试多台典型机组各装置出入口SO3浓度的情况。烟气经SCR 脱硝装置后,SO3的浓度均有不同程度的增加。经空预器后,有4%~8%的SO3被脱除。这是由于烟气温度的下降造成部分气相H2SO4会冷凝形成硫酸液滴,附着在金属壁面,造成腐蚀,同时烟气中的SO3也会与逃逸的氨结合生成硫酸氢铵堵塞空预器。进入电除尘装置,有12%~24%的SO3被脱除。由于电除尘装置的工作温度低于烟气的酸露点,因此会有部分SO3吸附在飞灰表面被脱除,也会有一部分SO3与飞灰中的碱性物质反应脱除。烟气进入脱硫装置后,有16%~30%的SO3被脱除。由于脱硫装置中烟气温度降至更低,气相H2SO4会冷凝形成硫酸液滴,但由于气相H2SO4粒径较小,脱硫装置对SO3脱除效率不高。Rhudy[24]研究发现湿法脱硫装置,对SO3脱除率一般在30%~50%。烟气通过湿式电除尘器进一步脱除,由于部分硫酸液滴(粒径在0.5~3μm)会形成亚微米气溶胶而无法脱除,最终造成PM2.5排放。

图5 燃煤电厂SO3迁移转化过程[22]
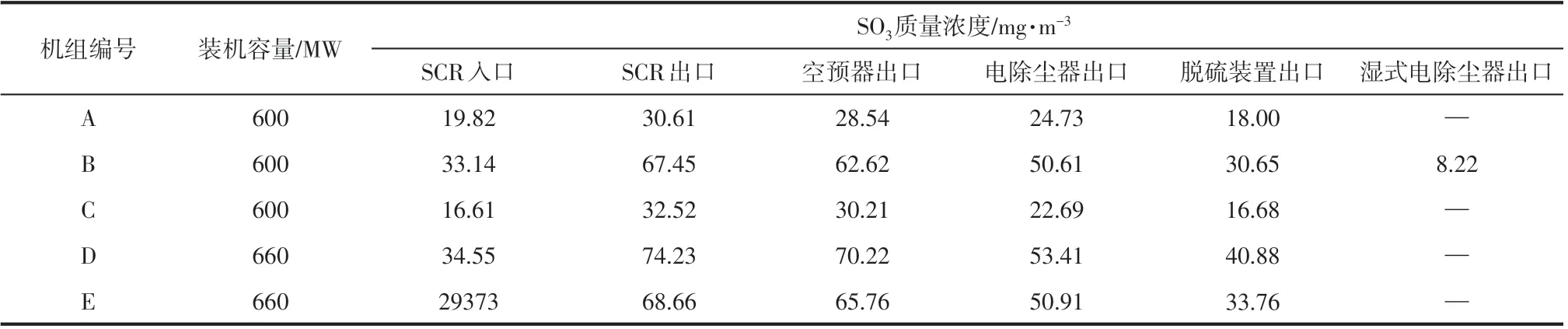
表1 SO3质量浓度测试结果
2.2 SCR脱硝过程中硫酸氢铵生成机理
目前,应用最为广泛的钒钛系SCR 脱硝催化剂的工作温度范围在320~400℃,因此燃煤电厂SCR脱硝反应器通常布置在锅炉省煤器和空预器之间。SCR脱硝系统中,氨等还原剂注入烟气中,与氮氧化物发生反应,生成N2和H2O[1]。但是,在SCR脱硝反应过程中,当喷入的氨过量或反应未能充分进行时,会有一定量氨随烟气离开SCR 脱硝系统,称为氨逃逸。逃逸的NH3会极易与烟气中的硫酸或SO3反应生成硫酸铵盐,如式(5)~式(8)[3]。
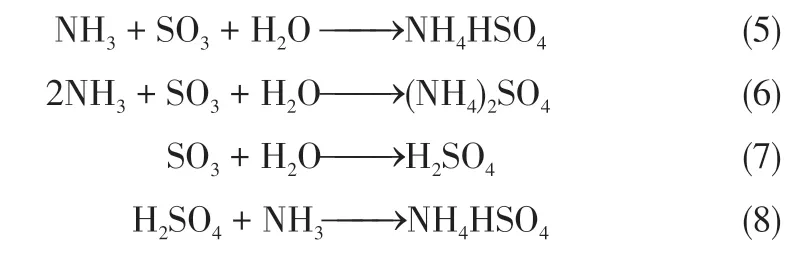
上述反应中,从热力学上分析会优先生成AS,而动力学分析却表明ABS 会优先生成,这是因为其反应速率更快[25]。图6 显示了NH3/SO3物质的量比对最终产物有着重要作用[3]。当氨体积浓度高于SO3时,硫酸铵将成为主要产物。随着NH3/SO3物质的量比的降低,产物为硫酸铵和硫酸氢铵。当这一比例进一步降低时,主要产品将是硫酸氢铵,而不是硫酸铵。当NH3/SO3物质的量比大于2 时,温度升高会促进硫酸氢铵的生成,因为硫酸铵在加热过程中会分解为NH3和硫酸氢铵。ABS的形成不仅取决于反应物的浓度,也取决于温度[26],ABS生成温度会随反应物的体积分数增加而增加。因此,ABS与AS 在不同温度和反应条件下,其生成机理的构建已成为了目前的研究热点。
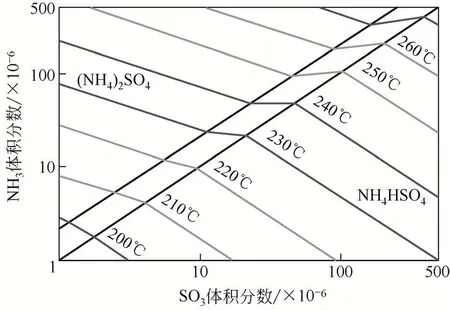
图6 硫酸铵和硫酸氢铵的形成[3,22]
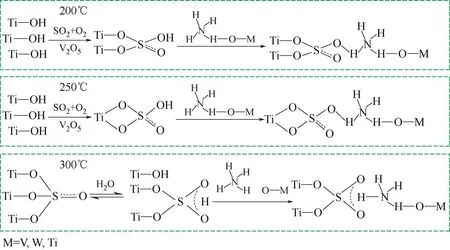
图7 催化剂上硫酸氢铵形成的机制[25]
Li等[28]研究了在不同温度下,硫酸氢铵在V/W/Ti催化剂上的生成和分解机理。如图7 所示,根据Lewis 酸碱理论,硫物种比钛物种具有更高的电负性,因此在硫酸化过程中容易形成Ti—O—S 带。弱吸附的SO2最初被活性钒位点氧化生成中间产物VOSO4,VOSO4在200℃、250℃和300℃下与TiO2结合分别形成了桥式双齿、螯合双齿以及三齿硫酸盐,这些硫酸盐与NH3反应生成硫酸氢铵。W的作用会降低硫酸氢铵的稳定性,硫酸氢铵会与WO3键合,而电子通过其中的键向硫酸氢铵的方向偏移,削弱了硫酸氢铵的稳定性,降低了其分解温度。NO和O2的存在可以破坏硫酸氢铵内部的键,并与硫酸氢铵发生反应,加速其分解,抑制其形成。
有学者采用理论和实验相结合研究方法,提出了硫酸氢铵在V2O5/TiO2催化剂上的形成、沉积和反应的综合机理[29]:硫酸氢铵主要在气相中通过SO3、H2O 和NH3的成核形成,然后沉积在催化剂表面。如图8所示,硫酸氢铵在V2O5/TiO2催化剂表面的分解包括两个步骤:通过硫酸氢铵的NH4+还原NO,形成N2和H2O,并将电子转移到相邻的钒位点,形成低价的V4+;然后通过O2或NO2将V4+氧化成V5+,实现V 的再氧化,这是决定硫酸氢铵分解反应速率的关键步骤。研究表明,NO2是比O2更好的再氧化剂。当NO2吸附在催化剂表面时,可以很容易从还原的V—OH—Ti 位点捕获H 原子,生成HNO2。在该反应之后,还原的V4+被再氧化为V5+,V/Ti 催化剂的结构和电子性质都得到有效恢复。
3 烟气中SO3以及ABS生成控制
3.1 喷射碱性物质控制SO3与ABS生成
目前研究中,可采用喷射碱性物质等方法控制SCR 脱硝系统SO3和ABS 生成。碱性吸收剂主要分为三类:钙基[CaO、Ca(OH)2、CaCO3],镁基[MgO、Mg(OH)2],钠基[Na2CO3、NaHCO3、NaHSO3]。在所有钙基吸附剂中,Ca(OH)2反应性最好,SO3去除效率在80%左右[30]。在350~400℃的温度范围内,Mg(OH)2比Ca(OH)2反应活性更好,且SO3去除率高达95%[31]。钠基吸附剂在燃煤电厂的实验中表现出了很高的去除效率。最常用的钠基吸附剂是Na2CO3、NaHSO3等。NaHSO3不能与烟气SO2进一步反应,因此其对SO3的选择性高于SO2。在物质的量比为1.0~1.5 时,SO3去除率达到95%以上[32]。吸附剂喷射形式也可分为干粉注射和浆液喷射,喷射的位置也可分为锅炉内、SCR装置和空预器。表2 对不同碱性吸附剂的优缺点进行了综合的比较[33-35]。
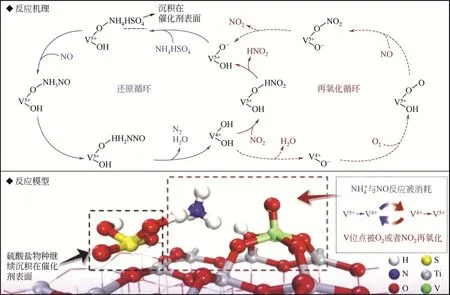
图8 硫酸氢铵在V2O5/TiO2催化剂上的形成和反应[26]
虽然上述方法能够从一定程度上对SO3进行脱除,但是对实际的脱除效率和脱除机理研究较少,对吸附剂中的有效成分、粒度、比表面积以及喷嘴的形状和布置方案等对SO3脱除的影响也需要进一步的研究。喷射碱性吸附剂并没有从源头上控制SO3以及ABS 生成,所以并不能很好地解决催化剂中毒以及空预器堵塞等问题。且喷入了碱性吸附剂之后,烟气中颗粒物的性质,如颗粒物的电阻、质量以及吸附性都会随之发生改变,所以需要进一步评估喷入碱性吸附剂对空预器以及ESP设备可能造成的影响。
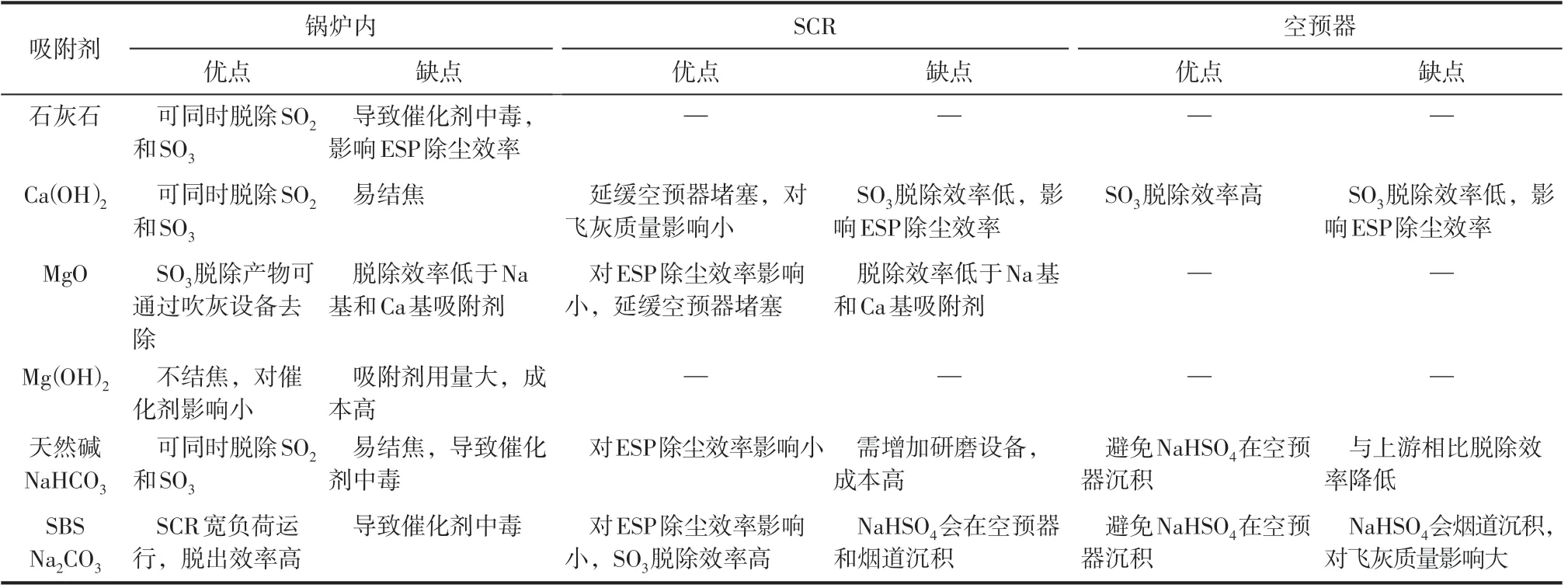
表2 不同碱性吸附剂的比较
3.2 催化剂活性组分对SO3以及ABS生成控制
开发低SO2氧化性能的SCR 催化剂也是控制SO3以及ABS 生成的研究热点之一。常见的SCR 催化剂包括贵金属型催化剂(Pt、Pd、Ag等)、过渡金属氧化物型催化剂(Mn、Fe、V、Cu、Cr、Co等)以及过渡金属离子交换型催化剂(Cu 和Fe)。金属氧化物催化剂在SCR 脱硝过程中应用广泛。目前应用最广泛的SCR 脱硝催化剂是以二氧化钛(TiO2)为载体、掺杂V2O5及WO3或MoO3的催化剂[36]。因为钒钛系SCR脱硝催化剂会将烟气中的一部分SO2氧化成SO3,同时催化剂表面容易形成硫酸氢铵造成催化剂失活,所以可以考虑对SCR 脱硝催化剂掺杂添加不同组分,以提高催化剂表面抗SO2氧化性能以及抑制硫酸氢铵生成。
3.2.1 活性组分W对催化剂的影响
金属氧化物WO3是V2O5/TiO2催化剂的典型促进剂,因为WO3催化剂在较低温度下表现出较强的抗SO2能力,也被越来越多受到学者的关注。Baltin等[37]发现在V2O5-WO3/TiO2催化剂上生成的ABS 可以在170℃下与NO 反应生成H2SO4,但在WO3如何促进的催化剂上硫酸氢铵分解等方面缺乏研究。之后Ye 等[38]发现含有WO3的催化剂表现出更强的抗SO2能力,WO3的引入促进了硫酸氢铵的分解。如图9所示,WO3的引入使SO2-4中S原子周围的电子云密度增加,这对硫酸氢铵中的+6价S原子还原为SO2中的+4 价S 原子有积极的影响,从而促进了硫酸氢铵的分解。
3.2.2 活性组分Co对催化剂的影响


图9 V/Ti和V-W/Ti催化剂上硫酸氢铵的分解[33]
3.2.3 活性组分Ce改性对催化剂的影响
除Co 元素外,Ce 基SCR 催化剂因其具有较高的储氧能力和优良的抗SO2氧化性能而一直备受关注[41]。Jin等[42]用溶胶凝胶法制备Mn-Ce/TiO2催化剂研究Ce 对催化剂上NH3低温还原NO 过程中抗SO2氧化性能影响。结果表明,Ce 改性催化剂表面覆盖的硫酸盐洗涤后,SCR活性基本能恢复。但对于不含Ce 的催化剂,其活性不能完全恢复。通过原位红外实验发现,Ce 掺杂能有效地抑制催化剂表面Lewis 酸位点的硫酸化,这使得吸附在Lewis 酸位上的NH3可以与气相NO 反应,提高了催化剂耐SO2的性能;实验同时也发现Mn-Ce/TiO2表面形成的硫酸盐比Mn/TiO2表面硫酸盐更容易分解,如图10 所示,这使得Mn-Ce/TiO2催化剂具有更好的耐硫性。
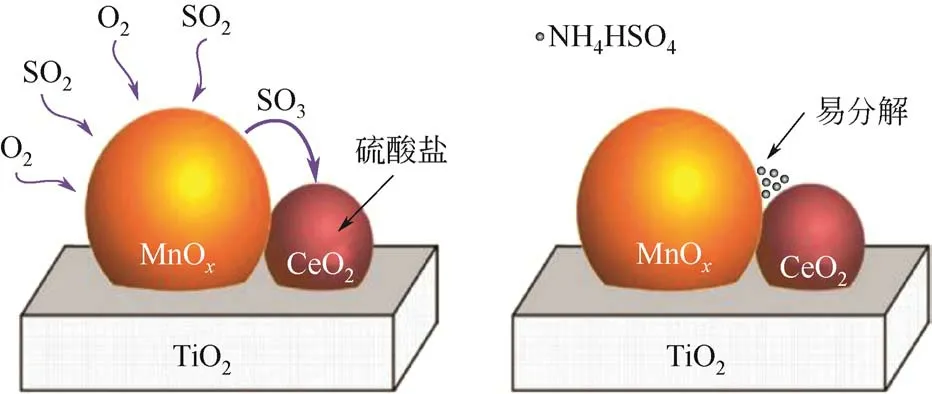
图10 Mn-Ce/TiO2样品上硫酸盐的形成途径[37]
3.2.4 活性组分Mo对催化剂的影响
研究表明,Mo 具有良好抗硫性能,已被证明是V2O5/TiO2基催化剂的有利添加剂。Kwon 等[43]研究Mo掺杂对V/Mo-Ti催化剂的增强性能和抗SO2性能的影响。结果发现,在钛酸中加入Mo 比在二氧化钛中加入Mo更有利于形成非晶态MoOx,添加的Mo会使NH3吸附密度和Brønsted酸性位点增加,这有利于提高催化剂性能。图11 显示了随着Mo6+比例增加,V/Mo-Ti 催化剂的活性增加,SO2抗性增大。这是由于Mo的加入抑制了SO2与V==O的反应,降低了SO2的吸附,从而抑制了硫酸铵盐的形成,提高了催化剂SO2的抗性。因此,与传统V/W/Ti催化剂相比,V/Mo-Ti催化剂具有更好的催化活性和SO2耐受性。
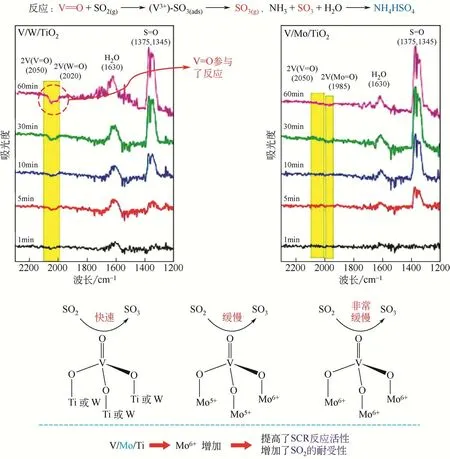
图11 V/Mo-Ti与V/W-Ti催化剂对比[38]
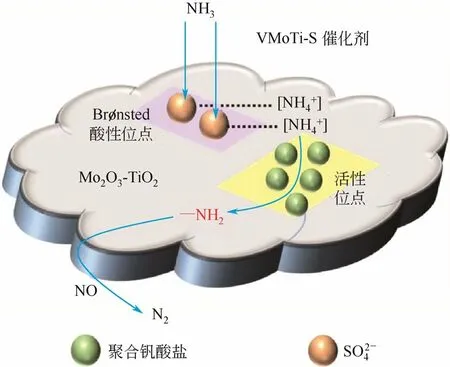
图12 V/Mo-Ti催化剂上的分离机制[39]
针对以上相关研究的分析可以发现,虽然通过选择合适的活性组分开发低SO2氧化性能的SCR催化剂可以从根本上解决目前SCR脱硝反应中SO3生成问题,但不同催化活性组分对催化剂上SO3和ABS生成影响机理目前尚不完善和清楚,相关研究亟待开展,以能为SO3和ABS的生成控制和燃煤机组的运行优化提供基础。
4 结语
(1)煤中硫含量和炉内氧浓度是影响炉内SO3生成的主要因素,此外SCR催化剂对SO2催化氧化是SO3的主要来源。SCR 脱硝系统中,烟气中NH3对气态SO3的生成有一定抑制作用。
(2)不同温度与不同NH3/SO3比例对最终ABS/AS产物起着重要作用。抑制SO3生成和降低氨逃逸是降低硫酸氢铵生成的关键,烟气中NO2能促进SCR催化剂表面硫酸氢铵一定程度的分解。
(3)采用碱性物质喷射是控制SCR 脱硝系统SO3和ABS 生成方法之一;同时,针对SCR 脱硝催化剂及反应过程,采用不同的活性组分对催化剂改性以及开发新型低SO2氧化SCR脱硝催化剂也是控制SO3和ABS 生成的有效方法,能够有效提高SCR反应活性以及抗毒性,这已成为目前SO3和ABS生成控制研究方向热点之一。
