Becker型肌营养不良合并扩张型心肌病的临床特征分析并文献复习
2021-04-20古帅鑫陈立杰
古帅鑫,陈立杰
Becker型肌营养不良症(Becker muscular dystrophy,BMD)是由DMD基因突变导致编码的dystrophin异常所致的X连锁隐性遗传病,因此男性多见,全球患病率为每10万名男性中1.53(95%CI0.26-8.94)[1]。BMD的临床症状与同样由于DMD基因变异引起的Duchenne肌营养不良(Duchenne muscular dystrophy,DMD)相似,但DMD中dystrophin几乎缺如,而BMD的dystrophin是分子量的改变或蛋白含量减少,由此骨骼肌症状往往较轻,也更具异质性[2]。DMD患者在童年时期就难以行走,十几岁开始依赖轮椅,在青少年时期继续失去上肢功能,并逐渐需要更多的辅助通气,心肌病通常在14至15岁时出现。尽管最近的研究心肺功能管理的改善正在延长患者的寿命,但早逝仍然通常在30岁时发生。一般来说,与DMD患者相比BMD发病较迟,平均发病时间为12岁,行动力的丧失被推迟到生命的第3个十年,心脏受累情况各不相同,并且整体预期寿命更长[3]。BMD患者心脏受累可能先于骨骼肌的衰退,心脏症状的发作和严重程度与骨骼肌病无关,是亚临床或轻度BMD患者的主要临床特征[4],但合并扩张型心肌病者较罕见。本文收集分析了1例合并扩张型心肌病的Becker型肌营养不良患者的临床资料并进行了随访,结果如下:
1 资料与方法
1.1 一般资料 患者男,33岁,河南人,因“四肢无力2 y,发作性胸闷、心慌1 y余”来我院就诊。2 y前无明显诱因出现四肢无力,伴肌肉疼痛,不能耐受长时间活动及重体力活动,无头晕头痛,无肌肉萎缩,无大小便障碍,未重视、未治疗。1 y前无明显诱因出现发作性胸闷、心慌,休息时好转,偶有间断剑突下不适,多于夜间及休息时出现、活动后加重。后就诊于我院心内科,查心脏彩超提示扩张型心肌病样改变:左心增大,二尖瓣轻度关闭不全,三尖瓣轻度关闭不全,左心功能下降(收缩+舒张)(左房40 mm,左室67 mm,EF值35%);动态心电图示:(1)基础心律为窦性心律,全程平均心率、最低心律及最高心率均在正常范围;(2)偶发室性早搏;(3)持续性ST-T改变,未见明显异常的动态变化;(4)心率变异率在正常范围内。诊断为:(1)扩张型心肌病:二尖瓣中度关闭不全 阵发性室速 心功能Ⅳ级;(2)腔隙性脑梗死;(3)鼻咽新生物;(4)左侧声带麻痹。给予“阿司匹林肠溶片、雅施达、倍他乐克、万爽力、能气郎、呋塞米、螺内酯”等药物治疗好转后出院。4 m前因阵发性胸闷、气喘,休息不能缓解,再次就诊于心内科,查心肌酶示:肌钙蛋白T 0.02 ng/ml;肌酸激酶1029 U/L;肌酸激酶同工酶24 U/L,经神经内科会诊后转入我科行肌肉活检,结果提示不排除肌营养不良,今为求进一步治疗再次来我科就诊。发病以来,神志清,精神可,饮食睡眠可,大小便正常,近1 y体重下降15 kg。既往1 y前因“声音嘶哑”就诊于我院,诊断为:(1)左侧声带麻痹;(2)鼻咽新生物。治疗好转后出院。家族史:家族中无与患者类似疾病,无家族遗传病史。入院体格检查:体温36.5 ℃;脉搏98次/min;呼吸24次/min;血压120/72 mmHg。
查体:神志清,精神可,自主体位,脑神经无异常,周身无肌肉萎缩及假性肌肥大,四肢肌张力降低,四肢肌力5级,双侧指鼻试验稳准,快复轮替试验正常,双侧跟膝胫试验稳准,Romberg征阴性,无不自主运动,步态正常,走“一”字正常,深浅感觉正常,生理反射存在,病理反射未引出。
1.2 入院后辅助检查
1.2.1 常规及实验室检查 肌酶谱提示:肌酸激酶761 U/L(参考值39~308 U/L),心肌损伤标志物:超敏肌钙蛋白T 0.019(0~0.1 ng/ml)、肌酸激酶同工酶 3.47(0~4.94 ng/ml)、肌红蛋白 43.58(0~100 ng/ml)、B型钠尿肽前体 46.59(0~97.3 pg/ml),既往肌酶及心肌损伤标志物变化见表1,血脂:高密度脂蛋白 0.84 mmol/L(>0.91 mmol/L),余未见异常。心脏彩超示:扩张型心肌病样改变,左心稍大,二尖瓣少量反流,左室功能下降(收缩+舒张)(左房径36 mm,左室径56 mm,EF值 45%),既往心脏彩超变化见表2;肝胆胰脾彩超:肝实质弥漫性回声改变(脂肪肝)。胸部CT:未见明显异常。普通心电图:正常范围心电图。双下肢MRI肌肉平扫示(见图1):双侧大腿外形基本对称,皮下及肌肉软组织形态信号未见明显异常。(1)双膝关节腔少量积液;(2)右侧胫骨前方软组织磁敏感伪影。

表1 肌酶及心肌损伤标志物变化(括号内为正常参考值范围)

表2 心脏彩超变化

图1 双腿肌肉平扫。T2 肌肉组织未见明显异常
1.2.2 肌肉病理活检 左侧肱二头肌病理活检提示(见图2):HE及GT染色肌束衣内结缔组织及脂肪组织稍增生,肌间小血管壁无增厚,管腔无狹窄,未见异常物质沉积,局部结缔组织内少量炎细胞浸润。肌朿内局部肌内衣稍増生,肌纤维大小不均匀,可见萎缩肌纤维,呈角形或圆形,散在分布,可见个别肌纤维坏死、吞噬,局部肌纤维核内移增加,可见固缩核团块,未见典型和不典型RRF。NADH和SDH染色显示少量肌纤维呈虫蚀样或靶样;NSE、PAS染色未见明显异常;ORO染色局部肌纤维内脂滴略增多。所见病理改变无明显特异性,但Dystrophin-N免疫组化呈阴性,请结合临床综合诊断并除外肌营养不良。
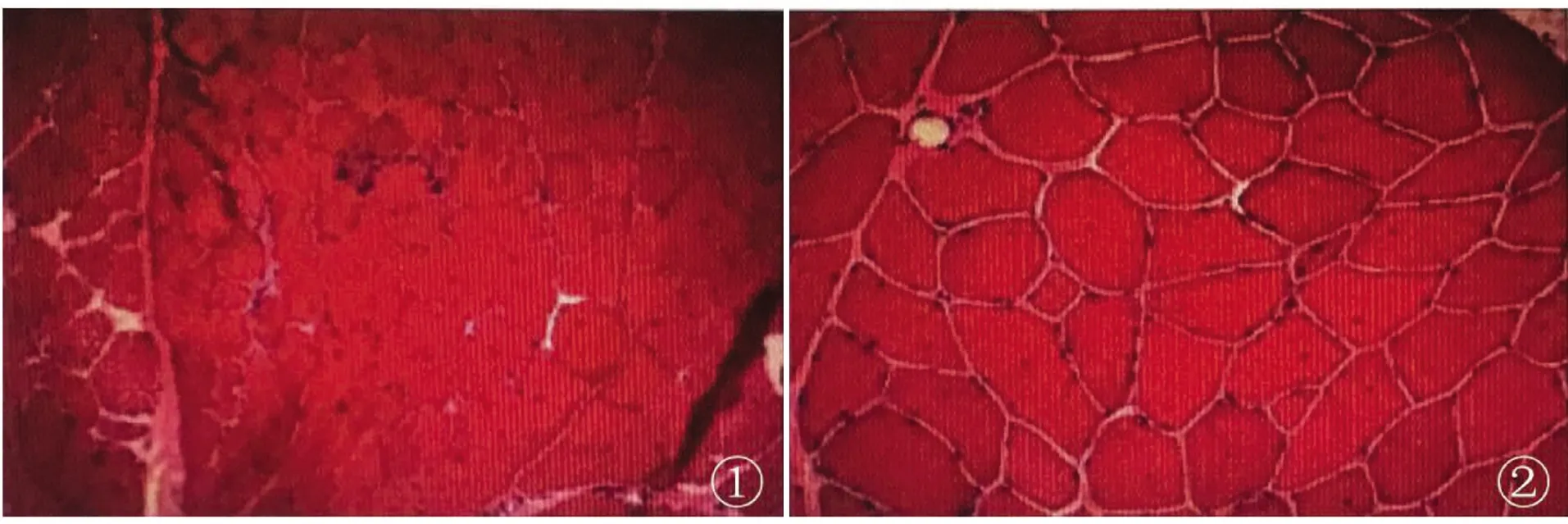
图2 左侧肱二头肌病理活检。HE染色(①×100,②×200)
免疫组化:Dystrophin-N(-),Dystrophin-R(膜+),α-Sarcoglycan(膜+),β-Sarcoglycan(膜+),γ-Sarcoglycan(膜弱+),δ-Sarcoglycan(膜+),Desmin(+),Spectrin(膜+),Utrophin(小血管周围+),CD3(个别细胞+),CD4(个别细胞+),CD8(个别细胞+),CD20(-),CD68(散在少量阳性细胞)。
1.2.3 基因检查 采用多重连接酶扩增反应技术结合毛细管电泳方法,分析了患者DMD基因79个外显子的缺失或重复情况,分析结果:患者DMD基因第10-18外显子缺失。分析意见:患者为DMD基因第10-18外显子缺失导致的贝氏肌营养不良。
1.2.4 治疗及随访 患者自2018年6月起便院外规律服用辅酶Q10、盐酸曲美他嗪片、ACEI,β-受体阻滞剂,醛固酮受体拮抗剂,利尿剂,伊伐布雷定片等维持治疗,随访至2020年7月16日,患者偶尔出现轻度肌肉酸痛、无力及咳嗽、胸闷症状,休息后可自行缓解,未再因心脏或肌肉症状加重而住院,日常可从事轻体力劳动,NYHA心功能Ⅱ级。
2 讨 论
DMD基因是人类基因组中最大的蛋白质编码基因,存在于X染色体上。该基因由79个外显子组成,主要在心肌,平滑肌和骨骼肌中表达[3]。DMD基因编码的dystrophin是一种位于肌纤维膜或肌膜下的低丰度蛋白,由如下四个主要结构域组成:将其锚定至细胞内肌动蛋白细胞骨架的氨基末端结构域,长而有弹性的杆结构域,将细胞内细胞骨架与细胞外基质连接的富含半胱氨酸的结构域,以及羧基末端[4]。该患者免疫组化染色提示 Dystrophin-N呈阴性,有研究表明位于N端基因的缺失可能会导致较早年龄发生心脏受累[5]。在功能上,dystrophin被认为作为抗dystrophin相关蛋白复合物(DAPC)的一部分,通过将收缩产生的力传递到细胞外基质而起到分子减震器的作用。DMD和BMD中经常观察到的认知障碍也可以通过神经元中dystrophin表达的改变来解释[3]。
DMD和BMD的损伤机制是多方面而复杂的。研究表明,膜损伤和钙离子内流是病理的关键,并得到了电子显微镜证据的支持,神经元型一氧化氮合酶和一氧化氮的损失以及活性氧和活性氮的增加也起着一定作用[6]。对具有导致DMD样表型基因突变的mdx小鼠进行的研究发现,肌肉收缩是发生初始肌肉损伤所必需的条件[7]。突变导致肌营养不良蛋白的功能受损使肌膜更脆弱并且更容易因肌肉收缩而损害,导致膜上产生小裂口。细胞外钙通过这些小裂口进入肌肉纤维,进而激活钙激活的蛋白酶,导致蛋白质降解[8]。心肌细胞中dystrophin蛋白的缺陷或缺失会通过类似的途径导致扩张型心肌病(Dilatedcardiomyopathy,DCM)。心肌纤维化开始于DMD的左室壁和BMD的右室壁,开始于心外膜,并进展到心内膜。它逐渐扩散到心室壁的大部分外半部。这种纤维化模式是肌营养不良症所特有的。心肌纤维化与年龄相关,且这种相关性不受肌营养不良类型的影响[9]。纤维化区将逐渐伸展,变薄,失去收缩性,并导致DCM。
未患病的骨骼肌是由横截面上大小相对均匀、呈多边形的纤维组成,相邻肌纤维之间几乎没有肌内结缔组织分隔。DMD肌肉以不同程度的萎缩、肥大、坏死、再生和肌内膜纤维化为特征,并伴有巨噬细胞浸润,淋巴细胞浸润则比较多变,在疾病后期,肌纤维在很大程度上被纤维脂肪组织所取代。在BMD中,骨骼肌病理与年龄相匹配的DMD患者相似,但较后者轻[10]。该患者肱二头肌病理活检结果与BMD肌肉病理相符,但病理改变无明显特异性,有研究报道,DMD和BMD整个病理变化的频谱还可以在各种基因型的先天性和肢带性肌营养不良中观察到[11],这表明肌肉病理对BMD诊断的价值有限。通过dystroplin抗体对肌肉组织进行免疫组化染色,可以观察到肌细胞膜上蛋白的表达情况,进而能够区分DMD、BMD和LCMD等不同类型,这对于临床表现,病理难以明确诊断的患者进行明确诊断,鉴别诊断,判断预后,正确地进行遗传咨询具有重要意义[12]。该患者最终通过免疫组化染色发现Dystrophin-N呈阴性,进而诊断为Becker型肌营养不良。该患者双下肢肌肉MRI检查未见明显异常,但有研究表明,在Becker肌营养不良症脂肪浸润之前,肌肉MRS可检测到PDE/ATP比值升高,这为BMD早期诊断提供了思路[13]。
DCM是一类既有遗传又有非遗传原因造成的复合型心肌病,以左室、右室或双心腔扩大和收缩功能障碍等为特征,通常经二维超声心动图诊断,临床常用诊断标准为左心室舒张期末内径(LVEDd)>5.0 cm(女性)和>5.5 cm(男性)[14]。该患者多次复查心脏彩超结果均符合扩张型心肌病的诊断。BNP水平是监测DCM进展的一个有价值的临床工具。在DMD所致扩心病中,BNP与射血分数、短轴缩短率或左心室内径等指标相关。随着心功能的恢复,该患者Pro-BNP逐渐下降并恢复至正常水平。BNP在DMD所致的DCM的心功能评价上可能也有着监测作用,但需要注意的是DMD/BMD的BNP水平低于特发性扩张型心肌病患者[15]。该患者乳酸脱氢酶,肌酸激酶同工酶多次测量均在正常范围内,故未列出,肌酸激酶始终高于正常值,这是由于肌膜通透性的增加使肌肉蛋白从肌纤维渗漏到血清中,只有随着肌肉质量的减少肌酸激酶水平才会下降。肌钙蛋白T连续监测始终高于正常值,这可能与心肌持续活动的特殊性有关,造成心肌细胞的持续性损伤,故对于肌钙蛋白持续升高,提示持续心肌损伤的患者,应考虑肌肉本身病变的可能。本例BMD患者动态心电图提示偶发室早,但随着纤维化的进展,可能会出现更严重的心律失常,包括心房颤动、房室传导阻滞、室性心动过速和心室颤动[4],因此应持续监测。
BMD目前仍然没有治愈的手段。DMD的标准治疗方案仍然是皮质类固醇激素,糖皮质激素可增加肌肉力量,延长独立运动,延缓心肌病的发作,改善肺功能并减少脊柱侧弯的发生率[16],在一项个案中还改善了伴有扩张型心肌病的BMD患者的心衰症状[17]。但糖皮质激素所带来的临床改善只是暂时的,并且还存在着体重增加,白内障和骨折在内的副作用[18]。基因治疗和细胞替代治疗能够纠正突变或替换骨骼肌和心肌中功能障碍或缺陷的蛋白,显得非常有前景。在DMD基因治疗方面,FDA已经批准了一种通过跳过外显子51来恢复开放阅读框架的突变特异性疗法,另外还有数项基因疗法的临床试验也正在开展中[2,19]。一项使用人脐带间充质干细胞移植治疗BMD家系成员的研究证明了该方法的安全性,和持续了12 w的治疗效益,虽然该实验缺少长期随访,但提供了一种潜在的治疗肌营养不良症的细胞疗法[20]。在DMD/BMD所导致的扩张型心肌病的治疗上,多项研究发现ACEI,β-受体阻滞剂等可改善DCM的心功能或减慢心肌纤维化的进程,且心力衰竭治疗的尽早开始可能会延迟左室功能障碍的进展[21~23]。该患者通过院外长期服用ACEI,β-受体阻滞剂等药物,心功能维持在NYHA心功能Ⅱ级,未进一步恶化,该患者服用的其他药物可能有效,但尚未有明确的证据支持。
总结及展望:以肢体无力为首发因素,且肌酶明显升高的男性扩张型心肌病患者,应考虑Becker型肌营养不良的可能。对于骨骼肌症状较轻的Becker型肌营养不良伴扩张型心肌病患者,心脏受累对患者的影响可能更大,ACEI和β-受体阻滞剂治疗有效,然而,随着患者年龄的增长和疾病的进展,病情仍然可能会恶化,基因治疗和细胞替代治疗的研究显得十分重要。
