儿童结节性硬化症1例报告并文献复习
2021-04-20孙平平马少春
孙平平,马少春
1 临床资料
患儿男,4月15天。因“反复抽搐5 d”于2018年8月8日入院。现病史:患儿5 d前无明显诱因出现抽搐,表现为点头拥抱样发作,多成串,每天1~2串,每串10余次,发作后精神差、睡眠多。近3 d发作频繁,每天3~5串。不伴发热、吐泻,吃奶量少,尿量减少。个人史:系G2P2,母孕期健康,按时产检均正常,足月顺产,出生体重3.1 kg,生后无窒息抢救史,母乳喂养。生后发育迟缓,目前竖头不稳,不会翻身,面部表情少,不会笑。家族史:父母体健,非近亲结婚,有1姐姐,体健,无抽搐及家族遗传病史。查体:生命体征稳定,神志清,前胸、后背、四肢皮肤可见散在多处色素脱失斑,最大者约2 cm×1 cm。腹部平软,肝右肋下2 cm,质软。四肢肌张力低,双侧Babinaki征阳性。辅助检查:血常规、血生化、免疫球蛋白及补体测定、淋巴细胞亚群6项、血沉、血氨、乳酸、β-羟丁酸、尿便常规均未见异常。脑电图示:各导联见多量广泛或多灶性中-高波幅不规则慢波夹杂棘波、多棘波、尖波短程阵发,两侧不同步、不对称,高度失律。颅脑MRI(见图1)示:双侧大脑半球皮质下白质T2信号稍高,双侧室管膜下见结节状短T1、短T2信号、FLAIR像呈稍高信号,提示多发结节改变。泌尿系超声:多囊肾。心脏超声未见明显异常。胸部CT:双肺下叶透光度不均匀,局部透光度增高,呈“马赛克”样改变,并可见磨玻璃样密度增高影。患儿家长未同意行眼底检查。综合分析,初步诊断为结节性硬化症(tuberous sclerosis complex,TSC)。因该疾病为基因变异所致,经家长同意,予完善TSC相关基因检测,发现受检者存在染色体16p13.3缺失(起始-结束位置2,113,376-2,147,267,大小为0.034 Mb),经两种软件分析发现,包含TSC2基因位置(chr16:2112488-2152981,12-42号外显子)存在杂合缺失变异(见图2),未发现受检者TSC1基因存在大片段变异。受检者父母及姐姐均未检测到TSC2基因存在大片段变异。结合基因检测结果,该患儿明确诊断为:TSC(2型)。治疗上给予甲泼尼龙冲击治疗3 d,后改为醋酸泼尼松片口服,同时口服托吡酯片控制癫痫,营养神经及对症支持治疗1 w,效果欠佳,仍有抽搐发作,每日1~2次,每次持续10~20 s,建议应用免疫抑制剂(雷帕霉素)治疗,家长拒绝。出院后继续口服托吡酯片及泼尼松片治疗。出院1 m、2 m后各电话随访一次,家长反应抽搐发作仍较多,每日1~3次,伴生长发育迟缓。
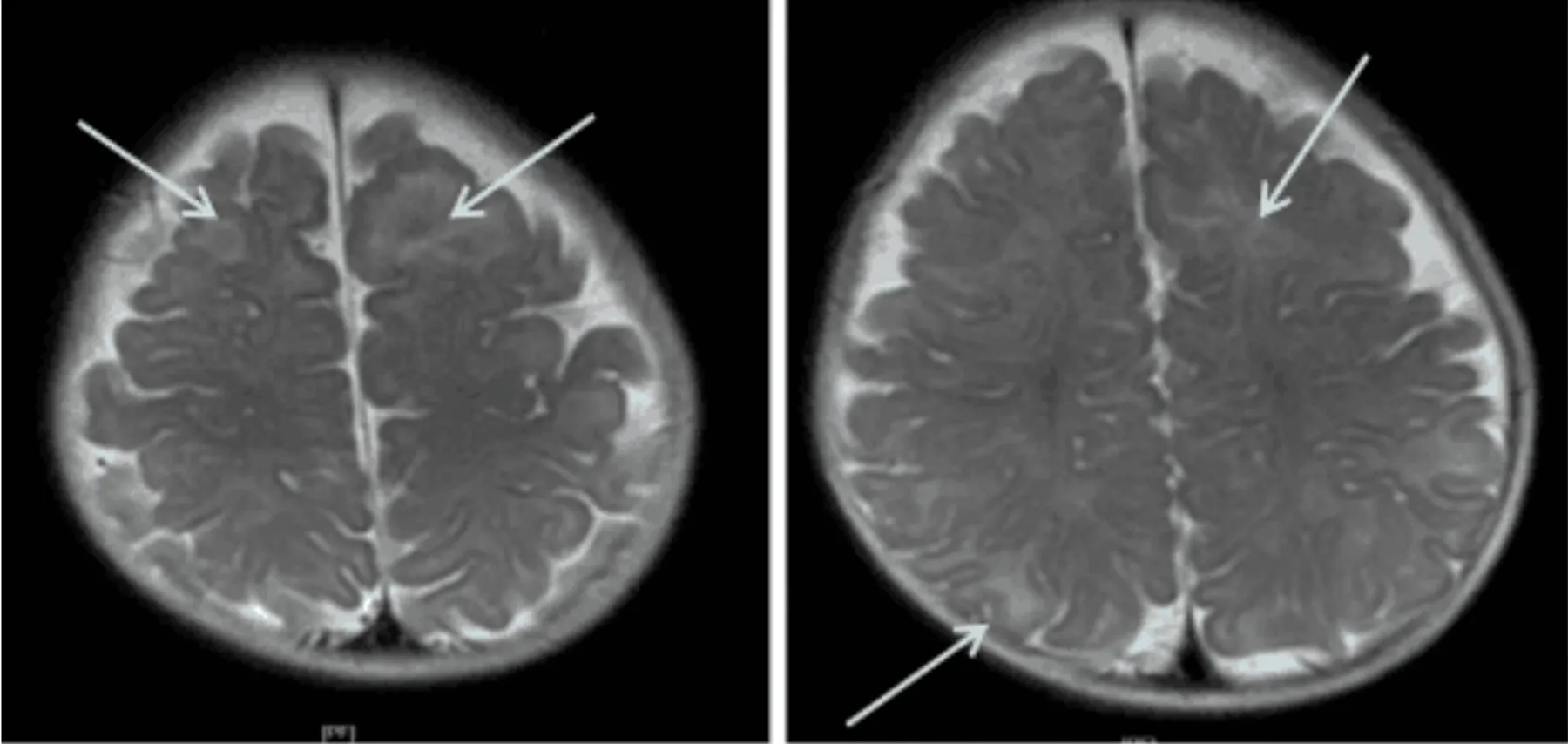
图1 双侧大脑半球皮质下白质T2信号稍高,双侧室管膜下见结节状短T1、短T2信号、FLAIR像呈稍高信号

图2 患儿TSC2基因12-42号外显子存在杂合缺失变异(注:黄色小球在纵坐标“1”位置代表正常,在“0.5”位置代表杂合缺失,在“0”位置代表纯合缺失)
2 讨 论
TSC的临床表现多样,主要有癫痫、智力低下、自闭症、色素脱失斑和牛奶咖啡斑、面部血管纤维瘤、鲨鱼皮斑、甲周纤维瘤、心脏横纹肌瘤、大脑皮质及室管膜下多发结节、血管平滑肌脂肪瘤、多发性肾囊肿等[1,2]。仅凭借临床表现有时很难确诊,甚至误诊或漏诊。随着医学技术的进步,基因检查成了该类疾病诊断的金标准。2012年国际TSC共识会议确立了TSC 的基因诊断标准,其可作为一个独立的诊断标准来确诊TSC[3]。目前已证实,TSC的致病基因有TSC1和TSC2两种,TSC1基因位于染色体9q34.3,TSC2基因位于染色体16p13.3,分别编码错构瘤蛋白和马铃薯球蛋白,这两种蛋白可在细胞中形成复合物,通过哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号通路调整细胞的生长,抑制细胞增殖。当基因发生突变时,会造成编码产物异常,抑制细胞增殖的作用减弱或消失,细胞过度增殖形成错构瘤,从而产生相应的症状和体征[4~6]。本例为4月龄婴儿,发育迟缓,反复抽搐发作,皮肤见多处色素脱失斑,脑电图高度失律,脑白质内和室管膜下多发结节,伴多囊肾,临床表现较典型,首先想到TSC,后经基因检测证实为TSC2基因杂合缺失变异导致的TSC(2型)。临床上大部分患儿表现不典型或表现轻微,这就需要借助于基因检查来明确诊断或鉴别诊断。但值得注意的是,目前仍有10%~25%的TSC患者检测不到致病基因,只能依靠临床表现诊断[3]。
新生儿TSC临床表现不典型,最常见的是皮肤改变,包括色素脱失斑、鲨鱼皮样斑、血管纤维瘤3种,其中色素脱失斑为最多见类型。心脏横纹肌瘤多在胎儿期被发现,也是生后就诊的原因之一。抽搐表现较少,其他表现更少,因此新生儿TSC容易漏诊或误诊[7]。陈湘湘等[8]总结分析6月龄以内婴儿TSC的临床特征,发现所有患儿头部MRI均可见多发性室管膜下结节,大部分同时合并脑皮质结节。以癫痫表现最多,其次为视网膜错构瘤、皮肤色素脱失斑、心脏横纹肌瘤、颜面部纤维瘤。梅道启等[1]对30例儿童TSC临床特点进行分析,共发现12种临床表现,其中以癫痫、智力低下、面部血管纤维瘤发生率最高。张林妹等[9]对105例小于18岁的TSC病例总结分析发现,色素脱失斑、癫痫发生率最高,肾脏病变发生率最低。国外学者Northrup等[3]也对TSC各器官病变的发生率做了统计,中枢神经系统病变包括:脑皮质发育不良90%,室管膜下结节65%~75%,室管膜下巨细胞星形细胞瘤5%~15%。本例患儿无抽搐家族史,母亲孕期体健,产检各项指标均未发现异常,故无法在胎儿期发现患TSC的风险。有文献报道,产前存在心脏横纹肌瘤的胎儿患TSC的风险为75%~80%,且多发性心脏横纹肌瘤的风险更高[4]。但该患儿病变未累及到心脏,所以不能在胎儿期做出预判。患儿皮肤多处色素脱失斑很容易在新生儿期被发现,但未引起家长和新生儿大夫的重视,未能及时就诊和诊断。住院后通过完善各项检查,发现患儿存在多器官系统受累,并且存在少见的肾囊肿病变。
TSC虽属于基因遗传病,但其散发病例数约占到2/3,提示其致病基因的自发突变率非常高。到目前为止,共发现了1117个TSC1基因突变位点和3221个TSC2基因突变位点,几乎涵盖了二者所有的外显子。TSC1和TSC2基因的致病突变类型多样,包括移码突变、无义突变、错义突变、剪切突变、缺失或插入,但二者均不存在突变热点[3,10]。TSC2基因包含的外显子较TSC1基因多,随机发生致病突变的概率高,二者的突变比例约为3:1,故TSC2基因突变可引起更加严重的临床表现[11]。通过对本例患儿及其父母进行基因检测,仅发现患儿TSC2基因区域存在大片段缺失变异,其父母无基因变异,提示该患儿属于基因自发突变发病。因患儿基因缺失片段包含31个外显子,故编码蛋白严重异常,会导致严重的临床表型。该患儿起病年龄早,生长发育迟缓,反复抽搐,病变累及到皮肤、中枢神经系统、肾脏、肺脏,口服抗癫痫药物治疗效果不佳,临床表型严重,与基因检测结果相符。Farach等[12]对92个TSC婴儿进行基因检测,发现63个存在TSC2基因变异,13个存在TSC1基因变异,16个未发现相关变异基因,基因总突变率为82.6%。其中TSC1基因变异包括移码突变(50%)、无义突变(42%)、剪切突变(8%)。TSC2基因变异包括移码突变(28%)、无义突变(23%)、错义突变(23%)、剪切突变(14%)、缺失(12%),可见缺失变异最少见。Boronat等[13]对280个TSC患者进行基因检测,160个发生TSC2基因变异,78个发生TSC1基因变异,42个未发现相关变异基因,基因总突变率为85.0%。国外研究中的基因突变率及突变类型与国内大致相同,但各突变类型所占比例存在一定差异[1,9],推测这可能与种族差异有关,或与国内缺乏大样本病例分析有关,希望通过深入研究分析来明确。
基于TSC患儿中癫痫发病率最高,对患儿影响最大,临床上的治疗方法主要有早期应用抗癫痫药物、mTOR抑制剂、生酮饮食、手术、迷走神经刺激术等。这些方法可有效控制癫痫发作,降低智力低下、癫痫性脑病、自闭症的发生率。mTOR抑制剂(如雷帕霉素)及其衍生物(依维莫司)通过阻止mTOR复合体1的激活和下游信号传导来发挥作用,是基于TSC发病机制的治疗药物,已被证实长期治疗TSC有一定的安全性,不良反应较小。在抗癫痫药物应用的基础上,联合应用mTOR抑制剂能缩小室管膜下星形细胞瘤和血管平滑肌脂肪瘤的体积,可改善患儿的临床症状[14]。根据最新证据,应考虑制定新的治疗指南,支持尽早启动mTOR抑制剂治疗,控制肾血管肌脂瘤,保护肾脏功能,防止未来严重并发症的发生[15]。本例患儿已口服抗癫痫药物,但仍不能很好的控制抽搐发作,并且患儿存在多器官系统受累,建议加用mTOR抑制剂治疗,家长未同意,经过2 m的随访,发现患儿病情无明显好转。
TSC的发病率相对较低,在临床工作中很容易被漏诊或误诊,特别是临床表现不典型的患儿。通过该病例,临床医生应提高针对TSC的诊断意识,对于发现心脏横纹肌瘤的胎儿、存在多处皮肤病变的新生儿、反复抽搐发作或智力低下的的婴幼儿,应进行全面的体格检查和完善辅助检查,尽早行基因检测,结合临床表现明确诊断。该疾病的主要治疗药物是抗癫痫药和mTOR抑制剂,并推荐尽早启动mTOR抑制剂的治疗,改善预后。
