疑难病研究:STAT3 基因突变致免疫失调综合征
2021-04-19姚安琪陈可可贺湘玲田鑫
姚安琪 陈可可 贺湘玲 田鑫
(湖南师范大学附属第一医院/湖南省人民医院儿童医学中心儿童血液肿瘤科,湖南长沙 410005)
免疫失调性疾病属于原发性免疫缺陷病的一种,是由于淋巴细胞增殖、调节性T 细胞功能异常及淋巴细胞凋亡障碍等引起免疫功能失调而导致的疾病,信号转导与转录激活因子3(STAT3)基因功能获得性突变可导致原发性免疫失调综合征[1-3]。该病以早发性自身免疫性疾病、反复感染为主要表现,首次报道于2014 年[2],迄今为止国内外相关文献报道多为个案或小样本资料研究[2-5],暂无发病率数据,临床医生对本病仍缺乏系统认识。目前国内外已报道病例43 例[3-4],其中国内报道仅1 例[3]。通过对所搜集病例的临床资料分析归纳,发现本病主要表现为早发性自身免疫性疾病及反复感染,其中以血液病、免疫缺陷、淋巴结增生最多见。本病发病早,无明显性别差异,目前尚无统一的诊断及治疗指南,在已报道的病例中多予以激素、免疫制剂或生物制剂进行治疗,亦有少数病例进行骨髓移植[4],但因样本数较少,尚不能对治疗效果进行统计学分析。本文报道1例STAT3基因功能获得性突变致免疫失调综合征,有利于提高临床医生对本病的全面认识,指导早期诊断及治疗。
1 病例介绍
病史:患儿,男,4 岁6 个月,因发热、咳嗽、面色苍白、乏力20 余天入院。患儿20 余天前无明显诱因出现发热、咳嗽,伴面色苍白、乏力,当地医院诊断为“支气管肺炎”。予以头孢他啶抗感染、雾化等治疗后热退、咳嗽好转,其间多次血常规示三系减少(中性粒细胞计数 0.01×109/L~0.26×109/L,血红蛋白 33~75 g/L,血小板计数41×109/L~335×109/L),因三系减少原因不明,故转我院进一步治疗。
既往史、出生史及家族史:既往体质欠佳,生后2 月龄开始出现反复呼吸道及消化道感染。患儿系第2 胎第2 产,足月顺产,出生体重2.5 kg,无生后缺氧窒息史,生长发育正常。父亲、母亲及哥哥身体健康,无类似家族史。
入院体检:体温 36.4℃,心率 108 次/min,呼吸 22 次/min,血压 90/50 mm Hg,经皮血氧饱和度 97%,身高 101 cm,体重 14.6 kg,贫血面容,全身皮肤黏膜无黄染、无皮疹。颈部及腋窝可触及数枚约0.5 cm×(0.5~1.0)cm 大小淋巴结,质韧,活动度可,无触痛。心肺体检无异常。腹平软,肝下缘在右锁骨中线肋下3.0 cm,质软,边锐,无触痛;脾下缘在左锁骨中线肋下2.0 cm,质软,边锐,无触痛。肠鸣音正常。神经系统体检无异常。
实验室检查:多次血常规示白细胞计数2.45×109/L~3.05×109/L( 参 考 值:3.69×109/L~9.16×109/L), 中 性 粒 细 胞 计 数 0.14×109/L~0.67×109/L(参 考 值:1.9×109/L~8.0×109/L),红细胞计数 1.69×1012/L~2.82×1012/L(参考值:3.68×1012/L~5.13×1012/L),血红蛋白 49~75 g/L(参考值:113~151g/L),网织红细胞计数 2.0×109/L~8.2×109/L(参考值:24×109/L~84×109/L),血 小 板 计 数13×109/L~95×109/L( 参 考 值:101×109/L~320×109/L)。淋 巴 细 胞 亚 群:NK细 胞 绝 对 值87.6/μL(参 考 值:230~801/μL),总T 淋巴细胞绝对值1 538/μL(参考值:2 284~4 776/μL),B 淋巴细胞绝对值812/μL(参考值:776~2 238/μL),淋巴细胞绝对值2 606/μL(参考值:3 320~7 006/μL),CD4CD8 双 阴 性T 细 胞 比例15.84%(参考值:0~1.5%)。C 反应蛋白、降钙素原、肝肾功能、心肌酶、电解质、血糖、凝血功能、甲状腺功能、狼疮全套、风湿抗体检查、血涂片、微量元素、血清铁蛋白、叶酸、维生素B12、地中海贫血基因、葡萄糖-6-磷酸脱氢酶、细胞因子、免疫球蛋白均正常。艾滋病毒抗原抗体、梅毒螺旋体抗体、乙肝病毒表面抗原、丙肝抗体定量、大小便常规、呼吸道病毒七项抗原、支原体抗体、EB 病毒抗体、新冠病毒核酸、TORCH、流感病毒抗原、中性粒细胞呼吸爆发试验均无异常。骨髓检查:骨髓增生不均一,大部分区域增生减低,粒系占62.5%,中幼粒及以下阶段可见,中性晚幼粒比值增高,形态大致正常,红系占1%,仅见中幼红细胞1%,形态大致正常,成熟红细胞大小不均,巨核细胞少见,血小板分布少。胸部CT:右下肺小气道病变。腹部彩超:肝上界6 肋间、肋下约33 mm,脾肋下约25 mm。
在全麻下行左侧腋窝淋巴结活检术,结果示淋巴结反应性增生。征得家长知情同意,采集患儿静脉血2 mL 进行全外显子组测序,发现STAT3基因第23 号外显子存在杂合突变c.C2147>T(p.T716M),即编码区第2 147 号核苷酸由胞嘧啶变异为胸腺嘧啶,导致第716 号氨基酸由苏氨酸变异为蛋氨酸,为错义突变。经一代测序验证确诊为STAT3基因杂合突变,患儿父母此位点均未见变异(图1)。根据美国医学遗传学与基因组学学会序列变异解读指南[6-7],该变异初步判定为疑似致病性变异(PS2+PM1+PM2+PP3):该患儿无家族史,经一代测序验证,为新发变异,符合PS2;该变异是位于变异热点和/或关键及既定的功能域的非良性变异位点,符合PM1;该变异为ESP 数据库(https://evs.gs.washington.edu/EVS/)、1000Genomes 数 据 库(https://www.internationalgenome.org/) 或ExAC 数 据 库(http://exac.broadinstitute.org)中正常对照人群中未发现的变异,符合PM2;多个生物信息学软件预测该变异会对基因或基因产物造成有害影响,符合PP3。
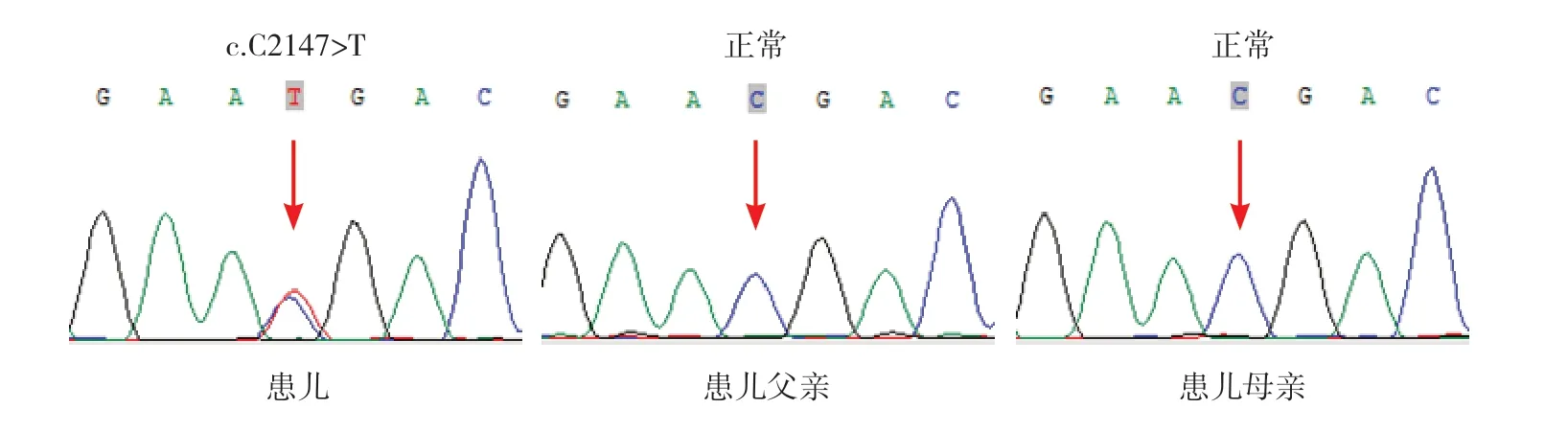
图1 患儿及其父母的STAT3 基因Sanger 测序图 患儿STAT3 基因存在致病性杂合变异c.C2147>T(p.T716M),为新发变异,其父母此位点未见变异。突变点如箭头所示。
2 临床经过
患儿入院后予成分输血、重组人粒细胞刺激因子和重组人促红素、头孢哌酮钠舒巴坦钠抗感染、雾化止咳等对症支持治疗,咳嗽基本好转,但肝脾较前继续增大(肝脏下缘在右锁骨中线肋下5.5 cm,脾脏下缘在左锁骨中线肋下3.5 cm),全血细胞减少无改善,完善骨髓穿刺排除血液系统恶性疾病后,考虑免疫性全血细胞减少,遂予甲泼尼龙(每日10 mg/kg,连用3 d)治疗。复查血常规示三系升高(中性粒细胞计数 2.78×109/L,血红蛋白 88 g/L,血小板计数 295×109/L),肝脾及淋巴结缩小后出院(此时基因结果尚未回报)。
出院后患儿未继续口服激素治疗,十余天后再次出现发热,复查血常规示三系减少,肝脾淋巴结再次增大。根据患儿自幼反复感染、全血细胞减少及肝脾淋巴结肿大等临床表现,结合基因检测结果,诊断为:免疫失调综合征(STAT3基因突变)。予以成分输血、头孢哌酮钠舒巴坦钠抗感染、口服醋酸泼尼松片(每日2 mg/kg),复查血常规示三系升高,肝脾及淋巴结缩小。出院后患儿规律口服醋酸泼尼松片(每日2 mg/kg),逐渐减量,现予1 片/日(每日0.3 mg/kg)。目前已随访8 个月,患儿偶有呼吸道或消化道感染表现,三系减少较前好转(中性粒细胞计数 2.37×109/L~8.57×109/L,血红蛋白 85~106 g/L,血小板计数92×109/L~347×109/L),浅表淋巴结不能触及,肝脾肿大明显缩小接近正常水平。
3 诊断思维
该患儿病例特点:(1)4 岁余男童,以发热、咳嗽、面色苍白及乏力起病;(2)肝脾、淋巴结增大;(3)自幼反复呼吸道及消化道感染;(4)反复多次全血细胞减少;NK 细胞绝对值、淋巴细胞绝对值及总T 细胞绝对值均降低,CD4CD8 双阴性T 细胞比例明显增高。
患儿表现为反复发热、肝脾和淋巴结肿大及全血细胞减少,可从感染或非感染性疾病两方面展开分析:(1)感染性疾病:在儿童发热并全血细胞减少病因中,感染性疾病占非造血系统疾病的50%,以病毒感染最多见,临床常见的病毒感染如巨细胞病毒、EB 病毒、艾滋病毒、微小病毒等均可引起[8]。患儿未检出相关病原,经抗感染治疗发热症状可好转,但全血细胞减少不能改善,故难以用感染解释。(2)非感染性疾病:①血液系统疾病:本例患儿为全血细胞减少,可排除单纯中性粒细胞减少症等一系血细胞减少的疾病。血清铁蛋白、叶酸、维生素B12等检查提示无造血物质缺乏,可排除营养性贫血。患儿无溶血性贫血相关的生化异常,不支持溶血性疾病。骨髓检查未发现幼稚细胞及病态造血现象,可与急性白血病及骨髓增生异常综合征等鉴别。患儿病程中无淋巴结进行性增大,不符合淋巴瘤的临床特征,淋巴结活检亦无恶性疾病证据,故可排除淋巴瘤。②结缔组织性疾病:风湿热、系统性红斑狼疮等结缔组织疾病可有反复发热,但患儿缺乏关节、肌肉、血管等结缔组织受累表现,炎症指标及血清铁蛋白不高,狼疮及风湿抗体检查未见异常,故不支持结缔组织性疾病。③免疫系统疾病:患儿自幼反复感染,考虑存在原发性免疫缺陷病。首先,患儿无B 淋巴细胞减少,免疫球蛋白正常,胸部CT 可见胸腺影,无卡式肺孢子菌等特殊病原反复感染的证据,无共济失调、毛细血管扩张及过敏性疾病等表现,亦无骨发育不良、维生素B12及叶酸代谢缺陷、高IgE 等特征性表现,故不支持联合免疫缺陷病、伴典型表现的联合免疫缺陷综合征、拟表型免疫性疾病或抗体缺陷病。其次,患儿虽有中性粒细胞减少,但并非出生后长期存在,骨髓检测未示髓系发育停滞,单核细胞及B淋巴细胞不低,故不支持吞噬细胞缺陷。补体检测正常,可排除补体缺陷。患儿无周期性发热、皮疹、关节炎等各部位炎症反应表现,可排除自身炎症性疾病。再次,患儿无分枝杆菌病、疣状表皮发育不良、单纯疱疹病毒脑炎等,排除天然免疫缺陷。患儿免疫球蛋白正常,无先天性畸形,不支持先天骨髓衰竭综合征[9]。患儿NK 细胞、淋巴细胞及总T 细胞计数均降低,CD4CD8 双阴性T细胞比例明显增高,需考虑免疫失调性疾病,本病主要表现为淋巴组织细胞增殖、自身免疫性疾病、反复感染。根据患儿临床表现要考虑该病,完善基因检测,结果提示STAT3基因功能致病性杂合突变c.C2147>T(p.T716M)。综上分析,患儿可诊断为免疫失调综合征(STAT3基因突变)。
4 诊断及确诊依据
诊断:免疫失调综合征(STAT3基因突变)。确诊依据:(1)临床表现:①患儿有发热、肝脾淋巴结肿大;②自幼反复呼吸道及消化道感染;(2)辅助检查:①反复多次全血细胞减少;②NK 细胞绝对值、淋巴细胞绝对值及总T 细胞绝对值均降低,CD4CD8 双阴性T 细胞比例明显增高;③基因检测示STAT3基因致病性杂合突变c.C2147>T(p.T716M)。
5 讨论
在2014 年的最新分类中,免疫失调性疾病被分为7 个亚类,包括家族性嗜血淋巴组织细胞增生症、调节性T 细胞病、自身免疫性淋巴细胞增生综合征等[9]。转录因子STAT 家族有7 个成员,STAT3 是其中之一,是多种代谢过程的关键调节因子,包括细胞增殖、存活、分化、调节自身免疫及炎症,因此STAT3突变可导致免疫缺陷、自身免疫或恶性肿瘤[4]。STAT3 的激活可能通过损害调解性T 细胞的发育,促进Th17 细胞的扩增和激活而导致自身免疫,因此可归于原发性免疫失调性疾病[2-3,10]。
STAT3基因功能获得性免疫突变致免疫失调性疾病平均发病年龄为3 岁,男女比率为0.82 : 1[4]。其临床表现表型多样,包括许多目标器官疾病,最常见的表现是自身免疫性细胞减少(68%,30/44)、淋巴细胞增生(66%,29/44)、肠病(55%,24/44)、间质性肺病(34%,15/44)、甲状腺炎(30%,13/44)、糖尿病(23%,10/44)[3-4]。其中,内分泌和胃肠道疾病发病最早,血液病次之,最迟出现的是间质性肺疾病。
该病临床表现:(1)血液系统:①血液病最常见(84%,37/44)[3-4],特别是自身免疫性细胞减少(30 例),包括自身免疫溶血性贫血、免疫性血小板减少性症及自身免疫性粒细胞减少,其中免疫性血小板减少性症最多见。②免疫缺陷:28 例(64%)有免疫缺陷表现,其中11 例反复呼吸道感染,18 例有低γ 球蛋白血症。11 例描述了调节性T 细胞水平,10 例表现为下降[4]。③淋巴增生:主要表现为淋巴结增大(57%,25/44)及肝脾增大(57%,25/44),其中21 例两种表现均有[3-4],另外3 例为纵隔腺病、1 例为嗜血细胞性淋巴组织细胞增多症。(2)消化系统:27 例(61%)出现肝脏及消化道症状[3-4],其中24 例发现肠病,临床主要表现为腹泻(18 例)、腹痛(4 例)及呕吐(2 例)。7 例肠病患者接受了靶向生物治疗(托西珠单抗、鲁索替尼等),其消化道症状及营养状况明显改善;另有2 例分别接受免疫抑制剂(硫唑嘌呤和他克莫司)治疗及干细胞移植,二者消化道症状均改善。9 例肝病患者,4 例进行肝移植后,1 例出现失弛缓症,3 例出现胰腺外分泌功能不全。(3)内分泌系统[4]:1 型糖尿病是本病最早的内分泌表现(23%,10/44),中位发病年龄为8 周。而甲状腺功能减退(30%,13/44)的中位发病年龄为2 岁。(4)呼吸系统:22 例(50%)表现为间质性肺病和复发性下呼吸道感染[4]。其中有3 例患者需要长期氧疗,1 例需要有创辅助通气。8 例患者接受免疫抑制剂治疗,2 例效果较好。1 例进行骨髓移植,但死于多器官功能衰竭[11]。3 例婴儿期表现为哮喘样症状的患者后来发现是间质性肺疾病,肺活检分别为弥漫性间质纤维化、弥漫性间质纤维化伴有肺泡增厚和巨噬细胞、不典型间质淋巴细胞浸润[12]。(5)生长发育:宫内生长受限5 例(11%),生后生长发育落后15 例(34%)。10 例患者进行胰岛素样生长因子(IGF-1)检测,7 例IGF-1 低[4],其中5 例患有肠病,可能因营养状况而影响IGF-1 水平。7 例患者接受生长激素治疗,3 例疗效好[5,11],4 例效果一般[2,4,11,13]。(6)其他[4]:皮肤病主要表现为特异性皮炎(27%,12/44)和脱发(9%,4/44)。9 例(20%)关节炎[14],6 例(14%)有骨质疏松,5 例(11%)有眼部疾病。3 例(7%)有肾脏疾病,1 例在成年时出现肾结石,1 例在5 岁时发生近端小管病变伴严重高钙尿,1例出现慢性肾功能损害且最终死于肾功能衰竭。
目前国内报道仅1 例[3],4 岁2 个月女童,主要表现为淋巴结肿大及全血细胞减少,反复多次呼吸道感染史,基因检测结果为STAT3基因第21号外显子存在杂合突变c.1974G>C(p.K658N)。该患儿前期不规范激素治疗,症状反复,后予以丙种球蛋白及规律激素治疗后症状好转,但其年龄小,随访时间不长,故不能评估长期疗效及预后。本例患儿临床表现及前期不规范激素治疗均与上述报道类似,后期规律激素治疗后即可获得较好疗效,但减量过程中出现反复,提示疾病仍有进展可能,故仍需长期随访观察。
治疗上尚无统一的专家共识或指南。多数医生使用激素和/或免疫抑制剂(包括环孢素、甲氨蝶呤、硫唑嘌呤、他克莫司等)治疗[4],总体疗效欠佳。英夫利西单抗、阿达木单抗、IL-1 单抗、CD20 单抗等生物制剂治疗亦有报道[3-4],但疗效均不令人满意。Milner 等[5]报道了1 位患者接受IL-6单抗治疗后其关节症状得到明显改善。Fabre 等[4]报道了7 例肠病患者接受托西珠单抗、鲁索替尼等治疗后其症状得到缓解。因此IL-6 单抗、JAK抑制剂是本病较有吸引力的治疗选择。造血干细胞移植是唯一可能根治该病的手段,但文献报道中5 例行造血干细胞移植的患者,其中4 例死于移植后并发症[4-5,11,13,15],提示移植时机、预处理方案选择及并发症的处理均有待探讨。
STAT3基因功能获得性突变致免疫失调综合征临床表现为各系统多重临床特征,部分患者既无多重自身免疫疾病也无血液系统疾病,易误诊或漏诊,临床医生需注意鉴别。目前本病暂无统一的诊断指南,基因诊断是唯一有效的诊断方法。
