孤儿核受体Nur77的多功能性及其相关药物靶点作用机制的研究进展
2021-04-15陈航姿
周 波,陈航姿,吴 乔
(厦门大学生命科学学院,细胞应激生物学国家重点实验室,福建 厦门 361102)
核受体是一类依赖于配体的转录调节因子,在细胞生长与分化,凋亡、自噬、焦亡等细胞死亡,机体代谢与发育以及稳态维持等方面发挥重要的功能.核受体功能的紊乱将导致一系列疾病的发生,如癌症、糖尿病、肥胖、不育、骨质疏松、阿尔茨海默病及心血管疾病等.核受体家族成员众多,包括类固醇激素受体、维甲酸受体、甲状腺激素受体、维生素D3受体及孤儿核受体[1-3].其中,孤儿核受体指无配体存在或暂时尚未发现其内源性配体的一类核受体.
核受体在机体的各器官组织中表达,与天然配体、激动剂或拮抗剂特异性结合后,可以发挥转录激活或转录抑制作用,因此,核受体被认为是一大类潜在的药物靶点[4].许多核受体的配体及能够激活核受体的药物被广泛应用于临床疾病治疗.例如:视黄酸受体(retinoic acid receptor,RAR)的配体全反式维甲酸,与As2O3联合使用对急性早幼粒白血病(acute promyelocytic leukemia,APL)具有良好的治疗效果[5-6];雌激素受体(estrogen receptor,ER)的拮抗剂雷洛昔芬(raloxifene)和他莫昔芬(tamoxifen)被广泛用于乳腺癌的治疗[7-9];噻唑烷二酮类药物(thiazolidinediones,TZD)作为过氧化物酶体增殖激活型受体(peroxisome proliferators activated receptors, PPARs)的配体,被应用于Ⅱ型糖尿病的治疗[10].因此,研究核受体相关的信号通路,寻找和开发以核受体为靶点的药物已成为当下热点和前沿.
核受体Nur77(也称TR3、NGFI-B 和NAK1) 是由立早基因NR4A1编码的蛋白,属于类固醇/甲状腺/维甲酸受体超家族的成员,结构上具有核受体的典型特征,但是其特异的内源性配体至今尚未被发现,因此被归类为孤儿核受体[11].NR4A家族包含Nur77(NR4A1)、Nurr1(NR4A2) 和Nor-1(NR4A3) 3个成员[12].Nur77在细胞增殖、分化、凋亡、机体代谢、免疫和发育等过程中发挥重要的作用.鉴于Nur77在不同生物学过程中可发挥关键的调控作用,筛选以Nur77为靶点的体外激动剂和拮抗剂显得尤为重要.
1 Nur77的分子结构特点
Nur77具有类固醇/甲状腺/维甲酸受体超家族的典型结构特征,由氨基端的转录激活域(transcription-activating domain,TAD)、中部的DNA 结合域(DNA-binding domain,DBD)以及羧基端的配体结合域(ligand-binding domain,LBD)组成(图1).其中,TAD是Nur77发挥转录激活作用的区域[13].DBD负责特异性识别单体Nur77 的应答元件(NGFI-B response element,NBRE)或者同源/异源二聚体形式的Nur77应答元件(Nur77 response element,NurRE),从而调控下游基因的转录;且DBD包含两个核定位信号(nuclear localization signal,NLS)序列,能够影响Nur77的亚细胞定位[14-15].虽然目前并未发现Nur77的内源性配体,但是其结构上仍具备LBD,且LBD还具有调控Nur77转录激活活性、二聚化形成和亚细胞定位的功能[14,16].

图1 Nur77结构示意图Fig.1 Schematic diagram of Nur77 structure
2 Nur77的调控方式
Nur77在不同的生物学过程中扮演着重要角色,其不仅作为转录因子调控下游靶基因的转录表达,还能作为调节因子通过蛋白间的相互作用调控其他蛋白的生物学功能.
2.1 基因转录调控
作为转录因子,Nur77主要通过其DBD与DNA应答元件结合,从而正调控或者负调控下游靶基因的转录和表达.Nur77不仅能以单体的形式与其应答元件NBRE结合[17](序列为AAAGGTCA),而且还能够以同源二聚体或者与其他家族成员(如Nurr1和Nor-1)形成异源二聚体的方式与应答元件结合[18-19](序列为TGATATTTACCTCCAAATGCCA).此外,在维甲酸受体的配体9-顺式视黄酸作用下,Nur77 还可以与视黄素X受体(retinoid X receptor,RXR)形成异源二聚体,与RXR的应答元件DR-5结合发挥转录激活的作用[20].Nur77对长链非编码RNA(long noncoding RNA,lncRNA)的转录也具有调控作用,如本课题组发现在肝癌细胞中,Nur77可以转录激活lncRNA WFDC21P,从而抑制肝癌的发生发展[21].
Nur77对下游靶基因的调控不仅由Nur77蛋白水平的高低所决定,也与Nur77结合应答元件的强弱及其募集辅因子的能力有关.例如:在表皮生长因子刺激下,Nur77蛋白水平被显著提高[22],从而激活下游靶基因的转录,包括调控细胞周期的基因CyclinD、E2F1等[23-24];凋亡诱导剂TPA的处理也能诱导Nur77蛋白水平的提高,激活Nur77下游凋亡相关基因的转录和表达[15,25-27].Nur77的翻译后修饰也会影响其转录激活活性.例如:在T淋巴细胞以及鼠成纤维细胞中,蛋白激酶B1(Akt1)通过磷酸化Nur77 DBD上的第351位丝氨酸,削弱Nur77与应答元件结合的能力,从而抑制其转录激活活性[28-29];而在肾上腺皮质激素(adrenocorticotropic hormone,ACTH)刺激下,位于Nur77 DBD上的第354位丝氨酸发生去磷酸化,进而促进Nur77与DNA结合,增强Nur77激活下游靶基因转录的能力[30].另外,Nur77的N端转录激活活性区域1(AF-1)可以通过募集辅激活因子如p300、SRCs、DIRP-205和PCAF等提高其转录激活活性[13].Nur77对辅抑制因子的募集则抑制其下游基因的转录.例如:在乳腺癌细胞中,Nur77能够募集辅抑制因子SWI/SNF复合体及HDAC1结合到脂肪酸受体基因(CD36)和脂肪酸结合蛋白4基因(FABP4)的启动子上,抑制两者的表达,进而抑制脂肪酸吸收[31];在肠癌细胞中,Nur77能够募集辅抑制因子HDAC3和TLE1,与转录因子TCF4相互作用,从而抑制下游靶基因c-Myc的表达,进而抑制肠癌细胞的增殖[32].
2.2 蛋白相互作用调控
Nur77除了作为转录因子调控下游靶基因的转录与表达,很多研究还表明Nur77可以通过与一些蛋白的相互作用而影响其生物学功能.首先,Nur77可以通过与转录因子结合影响它们的转录激活活性.例如:Nur77和糖皮质激素受体(glucocorticoid receptor,GR)结合可以互相抑制对方的转录激活活性;在视黄酸刺激下,Nur77通过与孤儿核受体鸡卵清蛋白上游启动子-转录因子(COUP-TF)竞争性结合到RXR上,可抑制COUP-TF与视黄酸响应元件(RARE)的结合,从而抑制下游靶基因的表达,进而抑制视黄酸诱导的细胞凋亡[33].本课题组也发现Nur77通过与β-连环蛋白(β-Cat)和T细胞因子4(TCF4)的相互作用抑制它们的转录激活活性,从而削弱Wnt信号通路的活性[32].Sun等[34]也发现Nur77与β-Cat的相互作用可以促进β-Cat降解,进而抑制β-Cat的活性.另外,Nur77通过与希佩尔-林道病肿瘤抑制蛋白(pVHL)的相互作用间接地抑制pVHL介导的缺氧诱导因子1α(HIF1α)泛素化降解,从而稳定HIF1α蛋白,促进血管内皮生长因子(VEGF)的表达和血管新生[35].

Chk2.细胞周期检测点激酶2;JNK1.c-Jun氨基末端激酶1;p38α.有丝分裂原活化蛋白激酶p38α;GSK3β.糖原合成酶激酶3β; CK2.酪蛋白激酶2;ERK.细胞外调节蛋白激酶;PKA.蛋白激酶A;RSK.核糖体S6蛋白激酶;MSK.有丝分裂原和 应激激活的蛋白激酶.红色为本课题组发现的磷酸化Nur77的位点,绿色为其他课题组发现的磷酸化Nur77的位点.图2 各种蛋白激酶磷酸化Nur77的不同位点Fig.2 Different sites of Nur77 phosphorylation induced by various kinases
其次,Nur77可以与细胞中蛋白激酶、乙酰化转移酶、甲基化转移酶以及代谢酶等多种酶相互作用进而调控酶活性.例如,Nur77的C端LBD能够介导Nur77与蛋白激酶Cθ(PKCθ)的结合,从而抑制PKCθ 的激酶活性,导致PKCθ介导的活化蛋白-1(AP-1)和核因子-κB(NF-κB)激活被抑制[36].本课题组的前期研究发现,p300的转录接头区域能够结合到Nur77 LBD上的FLELFIL序列;而Nur77的结合则覆盖p300的组蛋白乙酰基转移酶结构域,从而抑制p300的乙酰化转移酶活性,导致下游靶基因的转录被抑制[37].Nur77和甲基化转移酶PRMT1催化活性区域的相互作用可以抑制PRMT1对底物的甲基化[38].当肝癌细胞受到电离辐射时,Nur77可与DNA依赖蛋白激酶(DNA-PK)结合并被DNA-PK磷酸化,磷酸化的Nur77能提高p53磷酸化水平并增强其转录活性,促进电离辐射诱导的肝癌细胞凋亡[39].本课题组的研究还发现在血管紧张素Ⅱ(AngⅡ)诱导的心肌肥大过程中,Nur77的蛋白表达水平显著提高,并且在小鼠中敲除Nur77可以显著地削弱AngⅡ诱导的心肌肥大;进一步机制研究表明,Nur77能够通过与哺乳动物雷帕霉素靶蛋白复合体1(mTORC1)中的结节性硬化症蛋白(TSC1/2)结合形成三聚体,促进TSC2发生泛素化修饰,进而通过蛋白酶体降解,导致mTORC1激活;mTORC1的激活引起蛋白合成增加,细胞内活性氧(ROS)水平提高以及细胞体积增大,最终导致心肌肥大[40].另外,本课题组也发现Nur77能够与SUMO(small ubiquitin-like modifier)化(也称类泛素化)途径的泛素结合酶Ubc9竞争性结合磷酸烯醇式丙酮酸羧激酶1(PEPCK1),从而抑制PEPCK1的SUMO化修饰及降解[41].Nur77通过与脂肪酸氧化通路的硫解酶TPβ发生相互作用,影响其酶活性,进而调控脂肪酸氧化[42].
此外,Nur77可通过相互作用改变一些蛋白的构象,逆转这些蛋白的功能.线粒体定位的Nur77可以与B淋巴细胞瘤-2蛋白(Bcl-2)发生相互作用,导致Bcl-2的构象发生改变,暴露出其BH3结构域;该结构的变化使得Bcl-2无法再行使抑制凋亡诱导蛋白Bax和Bak的功能,反而抑制另一凋亡抑制蛋白Bcl-xL的功能,使Bcl-2的功能从抗凋亡蛋白逆转为促凋亡蛋白[15,43].
3 Nur77的翻译后修饰
常见的蛋白修饰包括磷酸化、乙酰化、泛素化、SUMO化、甲基化、氧化、糖基化等;此外,顺反式异构化和二磷酸腺苷(ADP)-核糖基化也是重要的蛋白修饰方式.Nur77蛋白的翻译后修饰对其发挥生物学功能具有重要影响,如转录激活、蛋白相互作用、亚细胞定位、蛋白稳定性等.
3.1 Nur77的磷酸化修饰
Nur77蛋白由598个氨基酸组成,其中含29个苏氨酸、69个丝氨酸和15个酪氨酸.如此密集的磷酸化氨基酸存在,提示Nur77很可能被不同的激酶磷酸化.组织细胞中Nur77的分子质量常在65~75 ku之间变化,这反映体内Nur77很有可能处于不同程度的磷酸化状态.在不同的刺激下,不同通路的激酶可以磷酸化Nur77的不同位点(图2),使得Nur77发挥不同的生物学功能.
首先,Nur77的磷酸化水平与其蛋白表达水平紧密相关.本课题组的一系列研究表明:Nur77的第95位丝氨酸被JNK1磷酸化后可以增强其与异构酶Pin1的结合,使Nur77发生异构化修饰,抑制Nur77降解,稳定Nur77蛋白[44];在化疗药物顺铂的刺激下,激活的Chk2能磷酸化Nur77的第88位苏氨酸,提高Nur77蛋白表达水平[45];而在茴香霉素刺激下,激活的JNK1能磷酸化Nur77的第95位丝氨酸,介导Nur77发生泛素化修饰,最终导致Nur77降解[46];当DNA发生双链断裂后,激活的DNA-PK能磷酸化Nur77的第164位丝氨酸,磷酸化的Nur77进而增强DNA-PK介导的p53磷酸化与转录活性[39].
其次,Nur77的磷酸化修饰能够调控其亚细胞定位.本课题组的研究发现:Akt1可以磷酸化Nur77的N端结构域,抑制Nur77与Bcl-2的相互作用,阻碍Nur77的线粒体定位,最终抑制细胞凋亡的发生[47];而且肿瘤细胞中高活性的Akt2可以磷酸化Nur77的第533位丝氨酸,使得Nur77滞留于细胞核,阻碍Nur77的线粒体定位[48];另外,在葡萄糖饥饿的情况下,激活的ERK2能够磷酸化Nur77的第237位丝氨酸,使得Nur77转位至线粒体,调节脂肪酸氧化[42].
再者,Nur77的磷酸化修饰可以调控其转录激活活性.如前所述,Nur77 DBD的第351位和第354位丝氨酸发生磷酸化修饰可显著抑制Nur77的转录激活活性,而Nur77 TAD的磷酸化修饰也对其转录激活活性有影响.本课题组发现,Chk2对Nur77的第88位苏氨酸磷酸化会抑制Nur77的转录激活活性,从而抑制Nur77下游抗凋亡基因的转录和表达,促进顺铂诱导的细胞凋亡[45].
此外,Nur77的磷酸化修饰会影响其蛋白相互作用.在巨噬细胞中,Nur77通过与p65(NF-κB的亚基)相互作用抑制NF-κB的活性;而在脂多糖(LPS)诱导下,激活的p38α可以磷酸化Nur77 的第27位和第143位苏氨酸,阻断Nur77与p65的相互作用,使Nur77丧失抑制NF-κB 信号通路的能力,导致NF-κB下游炎症因子激活[49].在肠癌组织中,活化的GSK3β 磷酸化Nur77的第27位和第143位苏氨酸以及第129位丝氨酸,可削弱Nur77与TCF4、β-Cat的结合,导致Nur77丧失对Wnt 信号通路的抑制作用,进而促进肠癌细胞的生长[32].
3.2 Nur77其他类型的修饰
Nur77除了发生磷酸化修饰外,还可以发生乙酰化、SUMO化、泛素化和氧化等修饰.本课题组的研究显示,在肝癌细胞HepG2中,乙酰化酶p300 可以使Nur77 LBD发生乙酰化修饰,抑制Nur77的泛素化修饰,从而稳定Nur77蛋白;而去乙酰化酶HDAC1则能降低Nur77的乙酰化水平及其蛋白表达水平[50].Nur77被JNK1磷酸化后会发生多泛素化修饰,通过蛋白酶体途径迅速降解;而E3泛素连接酶Smurf1能够介导Nur77发生非典型的泛素化,阻碍Nur77 的降解[51].Nur77的SUMO化修饰也能促进其发生多泛素化修饰,进而通过蛋白酶体途径降解[52].另外,在葡萄糖饥饿的情况下,Nur77的第505位、第551位和第566位半胱氨酸都会发生氧化修饰,进而调节脂肪酸氧化过程[42].
4 Nur77的生物学功能
Nur77几乎在人体各组织器官均有表达,其功能的紊乱将导致肥胖、糖尿病、心血管疾病、癌症等疾病.Nur77的生物学功能相当复杂,有的甚至截然相反.例如:Nur77既能促进肿瘤细胞增殖和存活,又能促进肿瘤细胞死亡;既能促进糖异生,又能促进糖酵解.Nur77的这些生物学功能依赖于外界不同的刺激而且具有组织细胞特异性.
4.1 Nur77与肿瘤
Nur77与肿瘤的发生发展密切相关.在不同的肿瘤组织中,Nur77表达水平存在差异,其中Nur77在黑色素瘤、结肠癌、胰腺癌、膀胱癌组织中高表达,而在肝癌、乳腺癌组织中低表达;而且Nur77在不同的肿瘤中发挥的功能有所不同,甚至在同种肿瘤的不同亚型或不同发展时期功能也有差异(表1).
本课题组发现在黑色素瘤中,高表达的Nur77对于黑色素瘤细胞适应葡萄糖饥饿的代谢应激状态至关重要[42]:在葡萄糖饥饿状态下,Nur77可维持细胞内还原型辅酶Ⅱ(NADPH)及还原型谷脱甘肽(GSH)的水平,从而降低葡萄糖饥饿引起的ROS水平升高,促进黑色素瘤细胞的存活.进一步研究发现,葡萄糖饥饿可诱导ERK2的激活并磷酸化Nur77的第237位丝氨酸,该位点的磷酸化作为分子开关,促进Nur77转运到线粒体,并且在葡萄糖饥饿状态下Nur77的线粒体定位是细胞存活所必需的,这与以往报道的Nur77在线粒体中促进细胞死亡不同.在线粒体中与Nur77相互作用的新蛋白是脂肪酸氧化最后一步反应的硫解酶TPβ,敲低TPβ或Nur77表达的细胞在葡萄糖饥饿状态下表现出类似的表型,如NADPH和GSH更容易丧失,而ROS升高更显著,说明TPβ可促进NADPH的产生而降低ROS水平.进一步机制研究发现,葡萄糖饥饿可诱导TPβ发生氧化修饰,从而抑制TPβ的酶活性,降低脂肪酸氧化,而在这一过程中Nur77比TPβ更容易被氧化,当它与TPβ结合后,通过自身氧化保护TPβ免于氧化引起的酶活性丧失,因而在葡萄糖饥饿状态下Nur77通过与TPβ结合促进NADPH和GSH的产生,降低ROS水平,使细胞得以存活.小鼠实验表明,Nur77和TPβ通过提高循环肿瘤细胞在体内的存活率,促进黑色素瘤的转移.临床样本统计分析也表明,Nur77在复发的黑色素瘤中表达水平明显高于原发的黑色素瘤.因此,Nur77通过保护脂肪酸氧化关键酶TPβ免于氧化失活,促进葡萄糖饥饿状态下的脂肪酸氧化,从而维持细胞中的ROS水平,最终促进黑色素瘤细胞的存活.
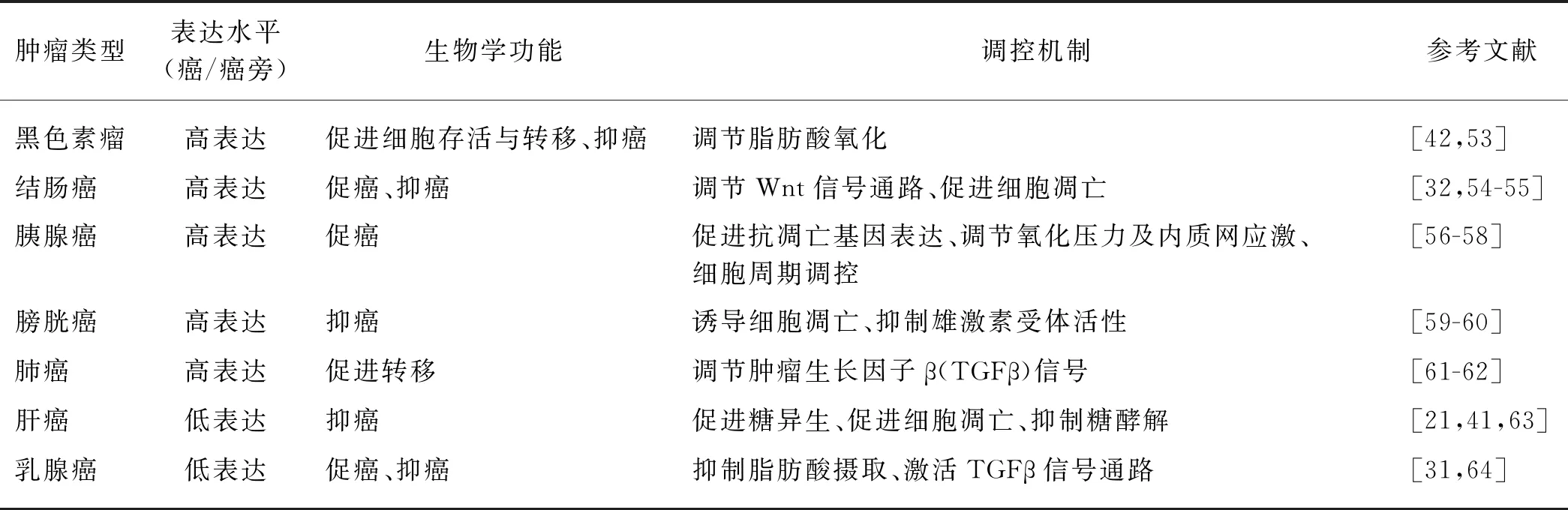
表1 Nur77在不同肿瘤中的作用Tab.1 The roles of Nur77 in different tumors
在结肠癌中,Nur77通过负调控Wnt信号通路活性,从而抑制结肠癌的发生[32]:一方面,Nur77通过与β-Cat相互作用,阻断其定位于Wnt下游基因的启动子;另一方面,Nur77与TCF4的结合进一步增强TCF4募集转录辅抑制因子的能力,进而抑制Wnt下游基因c-Myc和CyclinD1的表达,最终抑制肠道黏膜上皮细胞增殖和肿瘤的发生.在胰腺癌中,高表达的Nur77可以通过促进抗凋亡基因Bcl-2和Survivin的表达,调节细胞内氧化压力与内质网应激,并调控细胞周期来促进胰腺癌细胞的增殖与存活,最终促进胰腺癌的发生发展[56-58].在膀胱癌中,Nur77通过诱导细胞凋亡以及抑制雄激素受体活性,抑制肿瘤生长[59-60].在肺癌中,TGFβ激活的信号通路能够磷酸化Nur77并诱导其由细胞核转运至细胞浆,细胞浆中的Nur77与Axin2/Arkadia/环脂蛋白12(RNF12)形成活性复合体,诱导SMAD7蛋白通过蛋白酶体途径降解,从而促进肺癌细胞的侵袭和转移[61].
此外,本课题组通过临床样品检测和统计分析发现,随着肝癌的恶性程度增高,Nur77表达呈明显的下调趋势,且与肝癌患者的预后正相关;利用二乙基亚硝胺/四氯化碳(DEN/CCl4)和链脲菌素/高脂饮食(STZ/HFD)两种肝癌诱导小鼠模型(野生型和Nur77敲除小鼠),也证明Nur77能够显著抑制肝癌的发生发展[41].进一步机制研究表明Nur77通过抑制糖异生通路中限速酶PEPCK1 的SUMO化修饰,从而稳定其蛋白水平,最终促进糖异生、抑制糖酵解,阻断肝癌进程;但在肝癌发生发展过程中,Nur77启动子发生甲基化,导致其基因和蛋白表达下调,从而无法发挥抑制PEPCK1的SUMO化及其蛋白降解的功能[41].因此,抑制PEPCK1的SUMO化和提高Nur77的表达水平为肝癌治疗提供了一个新方向.在lncRNA研究中,本课题组发现肝癌细胞中Nur77能够通过转录激活lncRNA WFDC21P的方式调控糖酵解抑制肝癌的增殖和转移,最终抑制肝癌的进程[21].Nur77转录激活的WFDC21P能够与磷酸果糖激酶(PFKP)和M2型丙酮酸激酶(PKM2) 相互作用:一方面,WFDC21P通过与PFKP结合,抑制其四聚化,从而降低酶活性抑制糖酵解过程;另一方面,WFDC21P通过与PKM2结合,将其滞留在细胞浆中,阻碍其进入细胞核发挥转录因子的功能[21].本课题组早期研究还发现Nur77通过与转录因子p53结合,在转录和转录后水平调控癌蛋白小鼠双微体2(MDM2)表达,从而抑制肝癌细胞的生长[65].此外,Yang等[63]发现Nur77可以通过诱导肝癌细胞凋亡的方式发挥抗肿瘤的功能.
在乳腺癌的发生发展过程中,Nur77的功能目前并不明确.为此,本课题组首先在自发性乳腺癌小鼠模型(MMTV-PyVT)和化学试剂乙酸甲羟孕酮(MPA)-7,12-二甲基苯并蒽(DMBA)诱导的乳腺癌小鼠模型中发现,在两种小鼠模型中敲除Nur77都能促进乳腺癌的进展,而在Nur77敲除小鼠的乳腺组织中重新特异性地表达Nur77则可以显著抑制乳腺癌的进程;进一步机制研究表明Nur77在转录水平抑制CD36和FABP4的表达,阻断细胞对外源脂肪酸的吸收,从而抑制乳腺癌细胞的增殖[31].但Zhou等[66]认为Nur77能够通过激活TGFβ信号通路,促进乳腺癌的侵袭和转移.因此,Nur77在乳腺癌发生发展过程中发挥的功能及机制还需要更进一步的研究.
4.2 Nur77与肿瘤免疫治疗
肿瘤免疫治疗是近些年来兴起的肿瘤治疗方式.正常情况下,机体免疫系统可以识别并清除肿瘤微环境中的肿瘤细胞;但肿瘤细胞具备免疫逃逸的能力,能够采取不同的方式抑制机体的免疫系统,从而逃避免疫细胞的杀伤.T细胞遇到自身抗原、慢性感染或者暴露于肿瘤微环境后会发生功能紊乱,但导致T细胞功能失调的机制目前并不清楚.Liu等[67]在小鼠体外耐受性T细胞诱导系统中,对全基因组表观遗传学和基因表达测序,发现耐受性T细胞、效应性T细胞和调节性T细胞均具有显著差异,其中Nur77在耐受性T细胞中稳定性高表达;过表达Nur77则可以抑制效应性T细胞的分化,而在耐受性T细胞中敲除Nur77后,T细胞的耐受状态则被纠正,且对抗肿瘤和慢性病毒感染的能力增强.进一步机制研究表明,Nur77被优先招募到转录因子AP-1的结合位点,抑制AP-1的功能域,从而抑制下游效应基因的表达;Nur77结合之后可促进组蛋白H3K27ac的乙酰化,导致耐受相关基因的表达[67].同时,Chen等[68]也发现在嵌合抗原受体T细胞(CAR-T细胞)中敲除NR4A1家族能够获得更好的疗效,可大幅度延长荷瘤小鼠的生存期.此外,Karki等[69]报道表明Nur77拮抗剂Cl-OCH3能够抑制乳腺癌细胞PD-L1的表达,从而增强肿瘤免疫,抑制乳腺癌的生长和转移.因此,Nur77有望成为肿瘤免疫治疗的靶点.
4.3 Nur77与能量代谢
Nur77在机体能量代谢过程中发挥着重要的作用.Pei等[70]研究表明:在饥饿或胰高血糖素刺激下,小鼠肝脏中环磷酸腺苷(cAMP)信号通路能够提高Nur77的表达水平;在小鼠原代肝细胞和小鼠肝脏中过表达野生型Nur77,能够提高糖异生及糖转运相关基因的表达水平,如G6PC、FBP1/2、GPI1、烯醇化酶3基因(Enolase3)和葡萄糖转运蛋白2基因(GLUT2),促进葡萄糖的生成,提高机体血糖水平;而过表达Nur77显性负作用的突变体,则能够拮抗这些基因的表达,降低糖尿病db/db小鼠的血糖水平.而且在STZ诱导的小鼠和db/db小鼠的肝脏中Nur77表达上升,提示糖尿病小鼠中高血糖症状很可能是由肝脏中Nur77通过促进糖异生过程引起的.本课题组也发现Nur77能够调控糖异生通路的限速酶PEPCK1蛋白的稳定性来促进肝癌细胞的糖异生,进而抑制肝癌细胞的生长[41].
Nur77不仅在糖异生途径中发挥调控作用,在糖酵解、糖原分解和磷酸甘油的穿梭转运过程中也发挥了重要调控作用.如在骨骼肌细胞中,过表达Nur77能够促进许多糖代谢相关基因的表达,如糖原磷酸化酶基因(GP)、GLUT4和PFK等,从而促进骨骼肌细胞对葡萄糖的吸收和利用[71].
除了葡萄糖代谢外,Nur77也能调控脂类代谢.在小鼠肝脏中瞬时表达Nur77可以抑制固醇调节元件结合蛋白1-c(SREBP1-c)的表达,从而降低血脂含量和肝脏中甘油三酯的合成;在脂肪细胞的前体细胞中,过表达Nur77则可以通过缝隙连接蛋白1(GJA1)和Tolloid样蛋白1(TLL1)基因显著地抑制脂肪合成[72].Zhang等[73]研究也表明过表达Nur77能够使得脂肪细胞前体细胞处于静息状态,从而抑制脂肪合成,而敲除Nur77则可以促进脂肪细胞前体增殖以及增强脂肪合成能力.另外,Nur77还影响脂肪分解.Maxwell等[74]发现在骨骼肌细胞C2C12中,β-肾上腺素受体激活剂可以显著提高Nur77的表达,Nur77进一步促进脂肪分解途径中相关蛋白的表达,如CD36、解偶联蛋白3(UCP3)、单磷酸腺苷活化蛋白激酶γ3(AMPKγ3)、GLUT4和Caveolin-3等;而抑制Nur77的表达则显著削弱脂肪分解.在肝细胞中,Nur77通过抑制低密度脂蛋白受体和羟甲基戊二酰辅酶A还原酶的表达调控肝细胞中的胆固醇代谢[75].
此外,Nur77在线粒体代谢中也起到重要调控作用.在炎性巨噬细胞中,Nur77可以通过抑制异柠檬酸脱氢酶(IDH)的表达以及三羧酸循环的活力抑制炎症反应,最终抑制动脉粥样硬化等慢性炎症性疾病的发生[76].在黑色素瘤细胞中,Nur77通过与线粒体中硫解酶TPβ结合,调控线粒体中脂肪酸代谢过程,最终促进黑色素瘤细胞存活[42].
人类及其他哺乳动物能够通过维持能量摄取和能量消耗的动态平衡,长时间内保持体重稳定;但当摄取的能量超过自身能量需求时,将导致肥胖.目前,肥胖已经成为威胁人类健康的关键因素之一.瘦素由白色脂肪组织分泌,是一种抑制食欲的多肽类激素,作用于下丘脑,通过抑制摄食和增强能量消耗,在机体能量平衡中发挥重要作用.若机体无法响应瘦素,则引起肥胖[77].本课题组发现,Nur77的全身性敲除或下丘脑特异性敲低表达会导致小鼠对瘦素敏感性的显著下降[78].进一步机制研究表明,Nur77与瘦素下游关键信号分子信号传导与转录激活因子3(STAT3)能够发生相互作用,并且通过募集乙酰转移酶p300和促进去乙酰化酶HDAC1解离,提高STAT3的乙酰化水平,进而增强STAT3对下游基因的转录调控;随着小鼠年龄增长,Nur77表达水平低下与小鼠体内瘦素水平上升、摄食增多、血脂增高、能量消耗减少等密切相关,最终导致小鼠肥胖[78].该研究阐明了一条Nur77调控瘦素-STAT3的新信号通路,拓展了Nur77调控代谢的重要功能.由于肥胖患者机体对瘦素抵抗严重,Nur77能够通过增强瘦素信号通路缓解机体对瘦素的抗性,所以Nur77可作为一个潜在的靶点,用于开发预防和治疗瘦素抗性、肥胖等相关疾病的新型药物.
5 靶向Nur77的小分子化合物
Nur77的体内生理性配体至今未被发现.2003年Wang等[79]和Baker等[80]报道了果蝇(Drosophila)中Nur77家族成员Nurr1和Nur77同源物DHR38的晶体结构,发现在其配体结合口袋有6个疏水氨基酸,可能阻挡配体的结合.2005年PDB网站(http:∥www.rcsb.org)公布的晶体结构显示Nur77与其家族成员的结构类似,但到2008年尚未有直接结合并激活Nur77的化合物被报道.
5.1 第一个Nur77的体外配体Cytosporone-B
为了寻找Nur77的体外配体,本课题组以Nur77应答元件NurRE荧光素酶报告基因为筛选模型,从微生物内生真菌的天然产物库中筛选到一个可以显著和特异性激活Nur77转录激活活性的酮类化合物Cytosporone-B(Csn-B)[81].通过荧光滴定和圆二色光谱等生物物理方法证实Csn-B与Nur77有很强的结合能力,且特异性地结合在Nur77 LBD;进一步对Csn-B的生物学功能研究表明,Csn-B能通过Nur77 的介导提高小鼠的空腹血糖水平,且部分作用是通过促进Nur77对糖异生相关酶基因的转录调控实现的;Csn-B还能够诱导肿瘤细胞的Nur77转运出细胞核并定位于线粒体,导致细胞色素c由线粒体释放到细胞浆,进而诱导肿瘤细胞凋亡,最终抑制裸鼠移植瘤的生长[81].该研究首次发现Nur77激动剂,证实了孤儿核受体的体外配体存在,阐明有些孤儿核受体并非具有真正的配体非依赖性特征.此外,Csn-B也能抑制结肠癌、肝癌和乳腺癌的发生发展[21,32].
除了具有抑制肿瘤的效果外,Palumbo-Zerr等[82]研究发现Csn-B还可以治疗各种纤维化疾病,包括皮肤、肺、肾和肝纤维化等.该研究不仅佐证了Nur77介导Csn-B的功能和调控机制,而且展示了Csn-B很好的转化应用前景.在此基础上,本课题组以Csn-B为母体结构,进一步合成各种Csn-B衍生物,构建了特异靶向Nur77的小分子化合物库,从中发现了衍生物TMPA、THPN和PDNPA的独特功能.
5.2 靶向Nur77抑制小鼠血糖水平的小分子化合物TMPA
AMPK是机体重要的糖代谢调节蛋白,被认为是治疗Ⅱ型糖尿病的新药理学靶点.本课题组发现Nur77可以抑制AMPKα的磷酸化水平,却不影响其蛋白水平,提示还有其他因子参与抑制AMPKα磷酸化的过程[83].进一步研究表明AMPK的上游激酶LKB1在此过程中发挥重要的作用,Nur77通过与LKB1 结合,使其滞留在细胞核,抑制LKB1与AMPKα的结合,从而阻碍了LKB1对AMPKα的磷酸化;通过化合物筛选,发现小分子化合物TMPA能够直接结合Nur77,通过与LKB1竞争结合到Nur77的LBD,抑制Nur77与LKB1的结合,导致LKB1释放,进而磷酸化AMPKα,调控糖代谢;重要的是,在Ⅱ型糖尿病小鼠模型中,TMPA能显著降低小鼠的血糖水平,并改善小鼠对胰岛素的抵抗[83].该研究阐明了一条新的Nur77调控糖代谢的信号途径,即Nur77通过与LKB1相互作用调控糖代谢.LKB1作为AMPKα的重要上游激酶,直接调控AMPKα的降糖功能,在糖代谢中发挥重要作用[84-87].肝脏特异性敲除LKB1会导致严重的高血糖[84].因此,靶向Nur77/LKB1信号通路有望为治疗糖尿病的新药研发提供一个有效的筛选模型[83].
5.3 靶向Nur77诱导自噬性死亡的小分子化合物THPN
激活肿瘤细胞凋亡途径是当前临床治疗肿瘤的主要方法之一,但长期的药物使用导致肿瘤细胞对凋亡诱导产生抵抗,严重影响临床肿瘤治疗的效果.因此,寻找其他有效并可运用于临床的肿瘤治疗方式至关重要.营养缺乏时细胞通过自噬提供能量,维持自身存活;但过度自噬引起细胞器的过量清除则会导致细胞死亡.2014年本课题组报道了Nur77介导的线粒体自噬选择性地诱导细胞死亡,从而抑制黑色素瘤的发生发展[88].在小分子化合物THPN的诱导下,Nur77被Nix蛋白携带到线粒体,在线粒体外膜转位酶(TOM)复合体的帮助下进入内膜与腺嘌呤核苷酸转位酶1(ANT1)结合,使线粒体通透性转运孔道开放,引发线粒体膜去极化,导致大量的线粒体受损,继而诱发自噬对线粒体的过量清除,最终导致黑色素瘤细胞走向不可逆的自噬性死亡;小鼠模型进一步证实化合物THPN通过Nur77诱导的细胞自噬性死亡,能够很好地抑制黑色素瘤的发生发展和转移[88].该研究不仅阐明了Nur77通过线粒体信号通路参与细胞自噬性死亡诱导的新机制,还发现通过诱导细胞自噬性死亡能够克服黑色素瘤细胞对凋亡的普遍抗性,为黑色素瘤的临床治疗开辟了新思路.
然而,THPN对其他肿瘤却没有作用.为了进一步拓展THPN的应用,本课题组深入探讨了阻碍THPN诱导非黑色素瘤细胞自噬的根本原因和机制[48]:在许多非黑色素肿瘤细胞中激酶Akt2呈现高活性状态,通过磷酸化Nur77的第533位丝氨酸使得Nur77滞留于细胞核,阻断THPN诱导的胞浆Nur77与Nix结合,进而抑制Nur77线粒体定位及由此产生的线粒体功能受损,最终阻断细胞自噬性死亡.因此,THPN联合Akt抑制剂或者敲低Akt2表达就可以在细胞和动物水平上抑制不同类型的肿瘤生长.在此基础上,以THPN为母体结构,通过解析THPN-Nur77共结晶结构,进一步优化化合物结构,合成了不同的THPN衍生物,最终获得一个诱导自噬抑制肿瘤生长等性能高于THPN的化合物[48].该研究不仅确定了干扰THPN诱导肿瘤细胞自噬性死亡的具体分子,阐明了一条Nur77与Akt调控肿瘤耐药性的新途径,更为拓展THPN的应用范围和研发诱导自噬抑制肿瘤的新型药物提供了重要的理论基础.
5.4 靶向Nur77抑制脓毒症引起炎症死亡的小分子化合物PDNPA
脓毒症(即败血症)是感染后继发的一种全身炎性反应综合症,严重时导致死亡.本课题组首先在内毒素LPS及盲肠结扎穿刺(cecal ligation and puncture,CLP)诱导的小鼠脓毒症模型中发现,全身敲除Nur77的小鼠与野生型小鼠相比具有高敏感性和高死亡率,并伴随大量的炎症因子释放及组织器官严重损伤;进一步机制研究发现,Nur77抑炎功能通过与NF-κB亚基p65结合阻断其结合到DNA,由此抑制NF-κB转录活性以及下游炎症因子的激活,但在LPS刺激下,激活的p38α会磷酸化Nur77,使其丧失抑制NF-κB信号通路的能力[49].因此,阻断p38α与Nur77结合和磷酸化可以使Nur77更好地发挥功能.本课题组从自建的化合物库中筛选到以Nur77为靶点的小分子化合物PDNPA,其能够明显抑制NF-κB的转录功能,从而激活Nur77抑炎功能;共结晶结构及分子实验证实PDNPA能够结合到Nur77 LBD,阻断p38α的结合和Nur77磷酸化,使之发挥抑炎功能[49].该研究揭示了Nur77在脓毒症中发挥的新功能,阐明了其调控炎症的新途径,并发现了具有明显抑炎功能的化合物.
此外,厦门大学药学院张晓坤课题组2017年发现雷公藤素可以与Nur77结合,导致Nur77由细胞核转运至受损的线粒体,继而与肿瘤坏死因子受体相关因子2(TRAF2)相互作用而引起Nur77发生泛素化修饰,进而被溶酶体上的p62识别并通过细胞自噬清除受损的线粒体,最终达到抑炎效果[89].这一研究不仅为Nur77介导细胞自噬提供了新的依据,而且为Nur77调控炎症提供了新的理论支持.
除上述靶向Nur77研究外,Stephen课题组根据indole-3-carbinol代谢物也设计合成了一系列的小分子,包括C-DIMs(methylene-substituted diindolylmethanes)及其衍生物;这些小分子能够以Nur77依赖和非依赖的两条途径发挥抗肿瘤活性,既可以通过抑制Nur77的转录活性诱导细胞凋亡,也可以通过不依赖于Nur77的方式促进内质网应激诱导细胞凋亡[90].还有研究表明花生四烯酸[91]和前列腺素A2[92]能够结合Nur77 LBD,调控Nur77的转录活性.
6 总结与展望
Nur77通过调控靶基因转录、蛋白相互作用以及亚细胞定位等方式在不同的疾病中发挥着重要作用.首先,在不同的肿瘤或者同种肿瘤的不同亚型中,甚至同类型肿瘤发展的不同阶段,Nur77扮演的角色不尽相同,既能作为原癌基因发挥促癌作用,也能作为抑癌基因执行抑制肿瘤的功能;而且Nur77是抗肿瘤的重要药物靶点,抗肿瘤药物通过Nur77介导可以诱导细胞凋亡以及自噬性死亡而抑制肿瘤生长.其次,Nur77通过调控糖酵解、糖异生、糖原分解、脂代谢和线粒体代谢等生物学过程,参与机体的血糖以及能量平衡的调节.此外,Nur77在纤维化疾病以及炎症性疾病中也发挥重要调控作用.综上可见,Nur77在生命过程中发挥着多样且复杂的功能(图3).
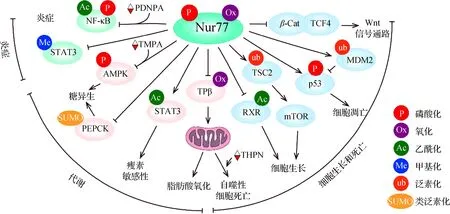
图3 Nur77的修饰、生物学功能及小分子化合物的作用机制Fig.3 Nur77 modification,biological functions and the mechanism of small molecule compounds
目前,临床肿瘤治疗的主要方式是手术切除、化疗法和免疫疗法.大部分的化疗药物通过诱导肿瘤细胞凋亡来抑制肿瘤的发生发展,长期使用导致许多肿瘤细胞产生凋亡耐受,限制了其进一步使用.因此,人们开始通过诱导肿瘤细胞新的死亡方式来筛选新的抗肿瘤药物.THPN通过Nur77诱导细胞过度自噬清除线粒体,最终导致黑色素瘤细胞走向不可逆的自噬性死亡,克服了黑色素瘤细胞对凋亡的抵抗[88],为治疗黑色素瘤和临床药物的筛选提供了新思路.
免疫疗法为癌症的治疗带来很大的希望,但是其应用有限,在一些实体瘤中发挥作用不够理想.T细胞是杀伤肿瘤细胞的主要执行者,但当T细胞暴露于肿瘤微环境之后会功能紊乱,呈现对肿瘤细胞耐受的状态;而Nur77在调控T细胞耐受的过程中发挥关键作用,敲除Nur77能够逆转T细胞的耐受状态[67],因此Nur77可能成为肿瘤免疫治疗的新靶点.以Nur77为靶点,继续筛选既能诱导肿瘤细胞死亡又能通过抑制Nur77的功能克服T细胞耐受状态,进而增强肿瘤免疫的药物,有望成为肿瘤免疫疗法的新方式,同时也能够为拓展免疫疗法的应用提供新思路.
Nur77是一个孤儿核受体,至今其内源性配体尚未发现,但本课题组通过深入系统的研究发现了一系列Nur77体外配体,能够结合到Nur77 LBD发挥调控作用,如通过诱导肿瘤细胞凋亡或者自噬性死亡抑制肿瘤、提高AMPKα磷酸化降低血糖,以及抑制NF-κB转录功能抑制炎症等.长期以来,人们主要筛选靶向核受体经典配体结合口袋并调控其转录激活活性的小分子,然而越来越多的研究证实核受体具有转录活性非依赖的生物学功能.本课题组的系列研究提示,针对核受体发挥功能的信号通路,筛选阻断或增强核受体与关键蛋白相互作用的小分子,有望成为筛选核受体靶向药物的新方向.
在核受体庞大的家族成员中目前一半左右的成员还是孤儿核受体,可能由于技术手段的限制,暂时没有分离和检测到它们的内源性配体.虽然体外激动剂与拮抗剂的发现为研究孤儿核受体的功能提供了强有力的工具,但是寻找和鉴定其真正的内源性配体仍然是一项重要和艰巨的研究工作.因此,发展新的分离技术和开创新的检测方法至关重要.
