原产地与入侵地椰子织蛾遗传分化特征
2021-04-13李优佳SHAMEER吕宝乾蒋方一丁章雨璐齐可欣
杨 帆, 李优佳, SHAMEER K S, 阎 伟, 吕宝乾, 蒋方一丁, 章雨璐, 涂 艳, 齐可欣
(1.海南大学林学院,海南 海口 570228;2.热带特色林木花卉遗传与种质创新教育部重点实验室,海南海口 570228;3.Insect Ecology & Ethology Laboratory, Department of Zoology, University of Calicut,Calicut 673635, India; 4.中国热带农业科学院椰子研究所,海南 文昌 571339;5.中国热带农业科学院环境与植物保护研究所,海南 海口 571101)
我国高温、高湿的热带地区自然气候条件十分优越,生物资源多种多样,为外来入侵物种的定殖、扩散和暴发提供了适宜的气候和食物条件[1-2].椰子织蛾(OpisinaarenosellaWalker)来源于印度和斯里兰卡,是近年来发现危害热带棕榈科植物的重要入侵食叶害虫.该虫最早于19世纪中期被发现,此后相继在缅甸、印度、孟加拉、印度尼西亚、泰国、马来西亚、巴基斯坦等地发现其危害[3-5];2013年入侵我国,现已扩散至海南、广西、福建和广东等地[6-8],严重影响椰子种植业的发展以及城市公共绿地的美观.鉴于其危害性,中华人民共和国国家林业局2014年正式把椰子织蛾列为危险性有害生物[9],2019年该虫被认定为三级危害性林业有害生物[10].国内外学者对椰子织蛾的研究主要集中在防治方面,有关其发生趋势的有效监测报道较少.明确椰子织蛾的入侵信息可有针对性地进行监测预警,从而有利于综合防控措施的实施[11],因此,准确把握该虫入侵来源是对其防控技术研发与应用的科学基础[12].
线粒体DNA因具有缺乏重组、突变率高、单倍体母系遗传等特点,被作为分子标记广泛应用于昆虫种群遗传变异和系统地理学的研究[13-14].学者已基于COⅠ基因分析了椰子织蛾不同地理种群的单倍型特征,其原产地与入侵地的单倍型存在一定的差异[15],但未系统深入分析.本研究利用COⅠ、COⅡ和COⅢ 3个基因片段分析原产地与入侵地椰子织蛾种群的遗传分化特征,以期为实施有效的检验检疫措施和监测其入侵动态提供理论依据.
1 材料与方法
1.1 样本来源
于2018—2019年在原产地(印度)和入侵地(中国、马来西亚、泰国和印度尼西亚)采集椰子织蛾样品,将采集的新鲜样品储存于无水乙醇中,-20 ℃冰箱保存备用.选取其中107份样品进行试验,其来源地见表1.

表1 原产地和入侵地椰子织蛾样品信息Table 1 Information of O.arenosella samples from the place of origin and invasive areas
1.2 基因组DNA提取及PCR扩增和测序
根据MagBeads Tissues Gen DNA Extraction Kit试剂盒说明书提取基因组DNA.所有样品的DNA质量在超微量紫外分光光度计下测定,D260 nm/D280 nm值均在1.7~2.0之间,将提取的DNA稀释至500 ng·μL-1后保存在-20 ℃冰箱备用.
根据椰子织蛾线粒体基因组的序列[16],分别设计COⅠ、COⅡ和COⅢ基因扩增片段的特异性引物,引物序列详见表2.各基因PCR反应体系为20 μL,包括2×Taq Plus Master Mix 10.0 μL、上游引物0.8 μL、下游引物0.8 μL、Template 1.0 μL (0.1 μg)、ddH2O 7.4 μL.PCR反应条件:95 ℃预变性5 min,95 ℃变性30 s,60 ℃退火30 s,72 ℃延伸1 min,15个循环(每个循环降1 ℃)之后95 ℃预变性30 s,50 ℃变性30 s,72 ℃退火60 s,28个循环,72 ℃延伸10 min,最后修复延伸10 min.PCR产物测序委托上海美吉生物医药科技有限公司完成.
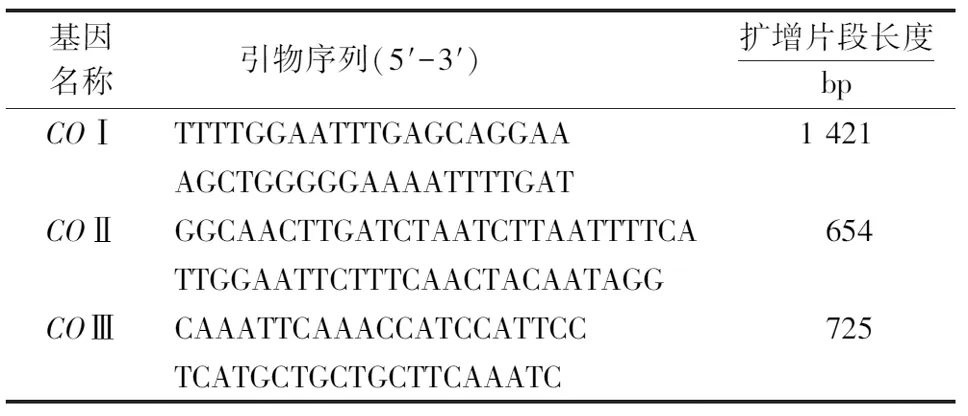
表2 椰子织蛾mtDNA-COⅠ、COⅡ和COⅢ基因片段引物Table 2 Primers for mtDNA-COⅠ、COⅡ and COⅢ gene fragments of O.arenosella
1.3 系统发育树的构建
利用BioEdit软件对3个基因序列进行比对和人工修正,去除两端低质量序列并区分杂合位点,以获得准确的基因片段序列[17].使用MEGA 6.0软件[18]分析序列的碱基组成和多态位点,并选取Kimura-2参数模型,以邻接(neighbor joining, NJ)法构建不同地理种群的系统进化树,系统树各分支的置信度用自举检验法(bootstrap)检验,共进行1 000次循环.利用PhyloSuite中的MrBayes[19]构建BI(Bayesian inference)树,依据ModelFinder[20]选择最佳替换模型为GTR+F.
2 结果与分析
通过基因序列比对分析,获得12个不同地理种群的107条COⅠ、COⅡ和COⅢ基因序列.经过拼接和引物删除,COⅠ、COⅡ和COⅢ基因序列片段长度分别为1 362、623和687 bp.从COⅠ基因序列鉴定出4种基因型(图1A),从COⅡ和COⅢ基因序列分别鉴定出3种基因型(图1B、1C).来自中国、马来西亚、泰国和印度尼西亚的椰子织蛾种群均为同一种基因型.
将COⅠ、COⅡ和COⅢ 3个基因片段序列拼接,序列比对显示其共包含35个变异位点,其中97%为碱基转换,3%为碱基颠换.在核苷酸组成上,原产地与入侵地种群的A、T、C和G的平均含量均分别为31.3%、41.7%、14.5%和12.5%,共存在34个变异位点,38%为嘌呤转换,62%为嘧啶转换.
将本研究测定的3个线粒体基因序列进行拼接,并以白胸织蛾(Endrosissarcitrella)COⅠ+COⅡ+COⅢ序列为外群,分别构建NJ系统发育树和贝叶斯树(图2).结果表明:所有入侵地的椰子织蛾基因序列聚成一支,支持率为100%;入侵地与原产地椰子织蛾种群形成两个明显的分支,遗传距离为1.3%(表3).
3 讨论
COⅠ、COⅡ、COⅢ是线粒体中最为保守的基因, 同源性为80%左右,所以其序列可作为研究物种系统进化和分类的有效分子标记[21].本研究选取COⅠ、COⅡ和COⅢ基因对原产地与入侵地椰子织蛾种群进行检测,A、T、C、G核苷酸的分布呈现非均一性,原产地与入侵地种群的各碱基占比相同.在35个核苷酸变异位点中,碱基转换发生的频率远高于颠换,该结果符合线粒体基因组进化规律.
本研究表明,入侵地区的椰子织蛾线粒体COⅠ、COⅡ和COⅢ基因与原产地种群基因的遗传距离为1.3%,低于普遍接受的种间遗传距离[13,22-24].系统发育树显示,椰子织蛾存在两个遗传支系,即原产地支系与入侵地支系,表明椰子织蛾种群已出现明显的遗传分化.如果这两个支系在形态和行为上出现差异,从进化的角度来说,可能发展为两个亚种.例如:与此类似的入侵害虫美国白蛾(Hyphantriacunea),在其原产地存在两种幼虫头壳色型[25];草地贪蛾(Spodopterafrugiperda)在原产地美洲长期进化过程中分化成两种生态型[26-27].
入侵地区椰子织蛾线粒体单倍型的多样性较低,而原产地种群则不然.这种低遗传多样性通常是由初始的少数个体直接造成的,这些初始种群在入侵的早期阶段会受到强烈的遗传漂变或奠基者效应的影响[28],种群经历的奠基者效应越多,其遗传多样性丧失越多[29].有研究认为该过程并非是不利的:一方面,通过遗传漂变和小种群中的非随机交配可以清除种群中的有害等位基因[30];另一方面,入侵地种群受环境选择压力的影响在新栖息地产生新的突变或杂交,可能预示这个物种侵占新领地的进化潜力.从寄主范围来看,椰子织蛾在其原产地主要危害椰子、海枣、董棕、油棕、野生枣椰、甘蓝椰子[31-32],入侵后,该虫还可危害多种景观树木,如酒瓶椰子、大王棕、银海枣、槟榔等[33-34],寄主范围进一步扩大.可见,椰子织蛾在与新环境中寄主植物的互作过程中产生了遗传变异和分化,以适应不同的环境,进而扩大了其分布范围.
入侵我国不同地区的椰子织蛾种群在线粒体基因组水平上未出现碱基变异,可能是由于其传入时间较短,尚未产生遗传分化.入侵我国的椰子织蛾与马来西亚、泰国以及印度尼西亚种群聚为一支,且它们具有完全一样的基因型,而该虫入侵马来西亚、泰国以及印度尼西亚的时间较我国早,推测入侵我国的椰子织蛾种群可能来源于这些国家.
