An improved scheme for infectious bursal disease virus genotype classification based on both genome-segments A and B
2021-04-12WANGYulongFANLinjinJIANGNanGAOLiLIKaiGAOYulongLIUChangjunCUIHongyuPANQingZHANGYanpingWANGXiaomeiQIXiaole
WANG Yu-long,FAN Lin-jin,JIANG Nan,GAO Li,LI Kai,GAO Yu-long,LIU Chang-junCUI Hong-yuPAN QingZHANG Yan-pingWANG Xiao-mei,,QI Xiao-le
1 Avian Immunosuppressive Diseases Division,State Key Laboratory of Veterinary Biotechnology,Harbin Veterinary Research Institute,Chinese Academy of Agricultural Sciences,Harbin 150069,P.R.China
2 OIE Reference Laboratory for Infectious Bursal Disease,Harbin Veterinary Research Institute,Chinese Academy of Agricultural Sciences,Harbin 150069,P.R.China
3 Jiangsu Co-innovation Center for Prevention and Control of Important Animal Infectious Disease and Zoonosis,Yangzhou 225009,P.R.China
Abstract Infectious bursal disease (IBD) is caused by infectious bursal disease virus (IBDV),which has a genome consisting of two segments of double-stranded linear RNA.IBDVs have been traditionally divided into four phenotypes based on their pathogenicity and antigenicity,including classic,variant,very virulent,and attenuated IBDV.With the emergences of divergent molecular characteristics of novel strains produced by continuous mutations and recombination,it is increasingly difficult to define new IBDV strains using the traditional descriptive classification method.The most common classification scheme for IBDV with segmented genome is based solely on segment A,while the significance of segment B has been largely neglected.In this study,an improved scheme for IBDV genotype classification based on the molecular characteristics of both VP2 (a viral capsid protein encoded by segment A) and VP1 (an RNA-dependent RNA polymerase protein encoded by segment B) was proposed for the first time.In this scheme,IBDV was classified into nine genogroups of A and five genogroups of B,respectively;the genogroup A2 was further divided into four lineages.The commonly used phenotypic classifications of classic,variant,very virulent,and attenuated IBDVs correspond to the A1B1,A2B1,A3B2,and A8B1 genotypes of the proposed classification scheme.The novel variant IBDVs including the strains identified in this study were classified as belonging to genotype A2dB1.The flexibility and versatility of this improved classification scheme will allow the unambiguous identification of existing and emerging IBDV strains,which will greatly facilitate molecular epidemiology studies of IBDV.
Keywords:infectious bursal disease virus,genotype,VP1,VP2,novel variant strain
1.Introduction
Infectious bursal disease (IBD),a major immunosuppressive disease of poultry,has been causing enormous economic losses to the poultry industry worldwide (Mülleret al.2003;Jackwood 2017).The causative agent of IBD is infectious bursal disease virus (IBDV),which is a variable doublestranded RNA virus (Heet al.2019).Two serotypes of IBDV(serotype I and serotype II) have been identified,but only serotype I strains are pathogenic to chickens (Mahgoubet al.2012;Qin and Zheng 2017).IBDV primarily targets the developing B cells in the bursa of Fabricius and causes massive loss of B cells (Kimet al.2000),which results in a decreased immune response and increased risk of secondary infection (Fanet al.2020).
IBDV is an icosahedral non-enveloped virus,which belongs to the genus Avibirnavirus of the family Birnaviridae.The genome of IBDV contains two segments,segments A and B (Brown and Skinner 1996;Mülleret al.2003).Segment A (approximately 3 200 bp) contains two open reading frames (ORFs).The smaller ORF encodes the non-structural protein VP5 (Rajaet al.2016),and the larger ORF encodes a polyprotein (pVP2-VP4-VP3),which can be cleaved by VP4 into the precursor pVP2 and VP3 (Petitet al.2000).VP2 is generated by further processing of pVP2,and then can interact with VP3 as a scaffold protein to assemble the capsid of IBDV (Saugaret al.2005).VP2 is the main host protective antigen and responsible for eliciting neutralizing antibody production (Deyet al.2019).A hypervariable region (HVR,aa 206–350) of VP2 has been identified where amino acid mutations are frequently observed (Letzelet al.2007;Jackwood 2017).This region is responsible for the virulence,antigenic variation,and cell tropism of IBDV (Brandtet al.2001;Jackwoodet al.2008;Qiet al.2009,2013,2016;Heet al.2019).Therefore,VP2 has generally been used for phylogenetic analysis.The RNA-dependent RNA polymerase VP1,encoded by segment B (approximately 2 800 bp),is essencial for the transcription and translation of the IBDV genome (Yeet al.2017).Segemnt B and the encoded VP1,also contribute to the virulence and evolution of IBDV (Escaffreet al.2013;Gaoet al.2014;Yanget al.2020).Recently,a phylogenetic marker for VP1 named B-marker (aa 110–252) was identified(Alfonso-Moraleset al.2015).
Traditionally,serotype I strains have been classified into four phenotypes according to their pathogenicity and antigenicity:classic IBDV (Cosgrove 1962),variant IBDV(Jackwood and Saif 1987),very virulent IBDV (vvIBDV)(Chettleet al.1989),and attenuated IBDV.With the emergence of novel strains of IBDV which have new molecular characteristics resulting from continuous mutation and recombination of the IBDV genome,categorizing new strains within the traditional descriptive classification system is becoming increasingly difficult (Jackwood 2012;Hernandezet al.2015;Lupiniet al.2016).Therefore,there is an urgent and pressing need for the development of a new classification method based on genotype for the molecular epidemiological study of IBDV.In 2017,one such genogroup classification scheme was proposed by Michel and Jackwood (2017) where serotype I strains were classified into seven genogroups based on the nucleotide sequences of the VP2 HVR,which was a significant breakthrough for the classification of IBDV.However,these seven genogroups cannot encompass the attenuated strains and the recently emerging novel variant strains of IBDV (Fanet al.2019;Xuet al.2019).In addition,as a segmented virus,segmentreassortment among different phenotypes of IBDV have been increasingly reported (Pikulaet al.2018;Hussainet al.2019;Pikułaet al.2020).Thus,while VP2 encoded by segment A is essential for the phylogenetic analysis of IBDV,an IBDV classification scheme based only on VP2 cannot provide a comprehensive accounting of all IBDV genogroups.It is indispensable to obtain information on both genome segments of IBDV for molecular epidemiology analysis.
In this study,an improved scheme for IBDV genotype classification was proposed that is based on both VP1(encoded by segment B) and VP2 (encoded by segment A).This approach is of great significance for the molecular epidemiological study of IBDV.
2.Materials and methods
2.1.Clinical samples
From July to November 2019,60 bursae were collected from domestic broiler flocks presenting suspected symptoms of novel variant IBDV infection as described in a previous report (Fanet al.2019).The bursal tissues were ground into 10% (w/v) homogenates in phosphate buffered saline(PBS,pH 7.2) with added penicillin and streptomycin.The homogenate was then frozen and thawed three times before centrifugation at 5 000×g for 5 min at 4°C.Finally,the supernatant was collected for further detection.
2.2.Viral RNA extraction and RT-PCR
Viral RNA was extracted from the bursal tissue homogenates using the Purelink RNA Mini Kit (Invitrogen,Carlsbad,CA,USA) according to the manufacturer’s instructions.The extracted RNA was reverse transcribed into cDNA using M-MLV reverse transcriptase (Invitrogen,Carlsbad,CA,USA).The PCR was performed using Prime STAR GXL DNA Polymerase (TaKaRa Biotechnology Co.,Ltd.,Dalian,China).To detect the viral genes,the forward primer P1 (5´-TCACCGTCCTCAGCTTAC-3´) and the reverse primer P2 (5´-TCAGGATTTGGGATCAGC-3´)were used to amplify a 643-bp fragment covering the HVR of the VP2 gene.A 1 513-bp fragment of the VP1 gene was amplified using the forward primer B206U(5´-AGAATGAGGAGTATGAGACCGA-3´) and the reverse primer B1718L (5´-GTCATCAATGGACCTCTC-3´).The PCR was performed under the following conditions:initial denaturation at 95°C for 5 min followed by 35 cycles at 95°C for 30 s,56°C for 30 s,and 72°C for 1 min,and a final extension at 72°C for 10 min.The RT-PCR products were detected by 1% agarose electrophoresis analysis.
2.3.Sequencing
The products of RT-PCR were purified using the AxyPrep DNA Gel Extraction Kit (Axygen,USA),and then sequenced by the Comate Biosciences Company (Changchun,China).The identified gene sequences were then submitted to GenBank.
2.4.Sequence analysis
Nucleotide sequences of the B-marker of VP1 and the HVR of VP2 were aligned with those of reference strains from GenBank (Appendices A and B) using the Clustal X Software(version 2.0) (Larkinet al.2007).Evolutionary analyses were conducted in MEGA6 (Tamuraet al.2013).The phylogenetic trees were constructed using the maximumlikelihood (ML) method with 1 000 replicates (Kimura 1980).Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Joining (NJ) and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach,and then selecting the topology with a superior log likelihood value.Molecular phylogenetic analysis was also performed using the NJ method based on the Kimura 2-parameter model (Kimura 1980).Finally,the ML tree was displayed with iTOL (Interactive Tree Of Life) (Beckerset al.2017).
3.Results
3.1.Detection of IBDV in clinical samples
The results of RT-PCR showed that the positive rate of bursal samples for IBDV was 35% (21/60).Subsequently,seven samples were randomly selected for sequencing the partial fragments of VP1 and VP2.The available nucleotide sequences of a 558-bp fragment (bp 547–1 104),corresponding to aa 183–368 encompassing the HVR (aa 206–350) of VP2,and a 1 252-bp fragment(bp 300–1 551),corresponding to aa 100–517 encompassing the B marker (aa 110–252) of VP1 of these seven IBDVs were determined and submitted to GenBank.The strain names and GenBank accession numbers of all seven IBDVs are listed in Appendices A and B.
3.2.Phylogenetic analysis of the HVR of VP2
Based on the nucleotide sequences of the VP2 HVR from the representative strains,the phylogenetic tree indicated that serotype I strains were clustered into eight genogroups(Appendix A;Fig.1).In addition to the seven previously identified genogroups (genogroup A1–A7) proposed by Michel and Jackwood (2017),the attenuated strains of IBDV formed a new branch,named as genogroup A8 (Fig.1).In addition,serotype II strains were classified into genogroup AII.Furthermore,the genogroup A2 was further divided into four lineages of A2a,A2b,A2c,and A2d.The recently occurring novel variant IBDVs (Fanet al.2019) formed the new lineage of A2d (Fig.1;Table 1).All seven strains isolated in this study shared a comparatively higher identity(98.6–100%) and the same conserved amino acid residues(217K,252I,and 299S) with the reference strains of novel variant IBDV of SHG19,SHG120,and SHG350 (Fanet al.2019).In total,IBDVs were classified into nine genogroups based on the VP2 HVR,and all IBDV strains identified in this study were clustered into genogroup A2d of novel variant IBDVs (Fig.1).
3.3.Phylogenetic analysis of representative fragments of VP1
The maximum-likelihood tree based on the nucleotide sequences of the B-marker of VP1 from the representative strains (Appendix B) showed that serotype I strains formed four major genogroups (Appendix B;Fig.2).Genogroup B1 consisted of classic,attenuated,and early variant strains.The recently reported novel variant strains from China (Fanet al.2019) and Algeria reassortant strains (Abedet al.2018) were also classified into genogroup B1.Genogroup B2 was composed of vvIBDV strains.Genogroup B3 strains were HLJ0504-like strains with segment B from a unique ancestor (Qiet al.2011;Heet al.2014;Hussainet al.2019,2020).The emerging transitional-lineage strains in Poland and Finland (Tammirantaet al.2018;Pikułaet al.2020)formed genogroup B4.Similar to the classification of VP2,serotype II strains were classified into genogroup BII.In short,IBDVs were classified into five genogroups based on the B-marker of VP1,and all IBDV strains identified in this study were clustered into genogroup B1 (Fig.2).
3.4.Novel genotype classification of IBDV
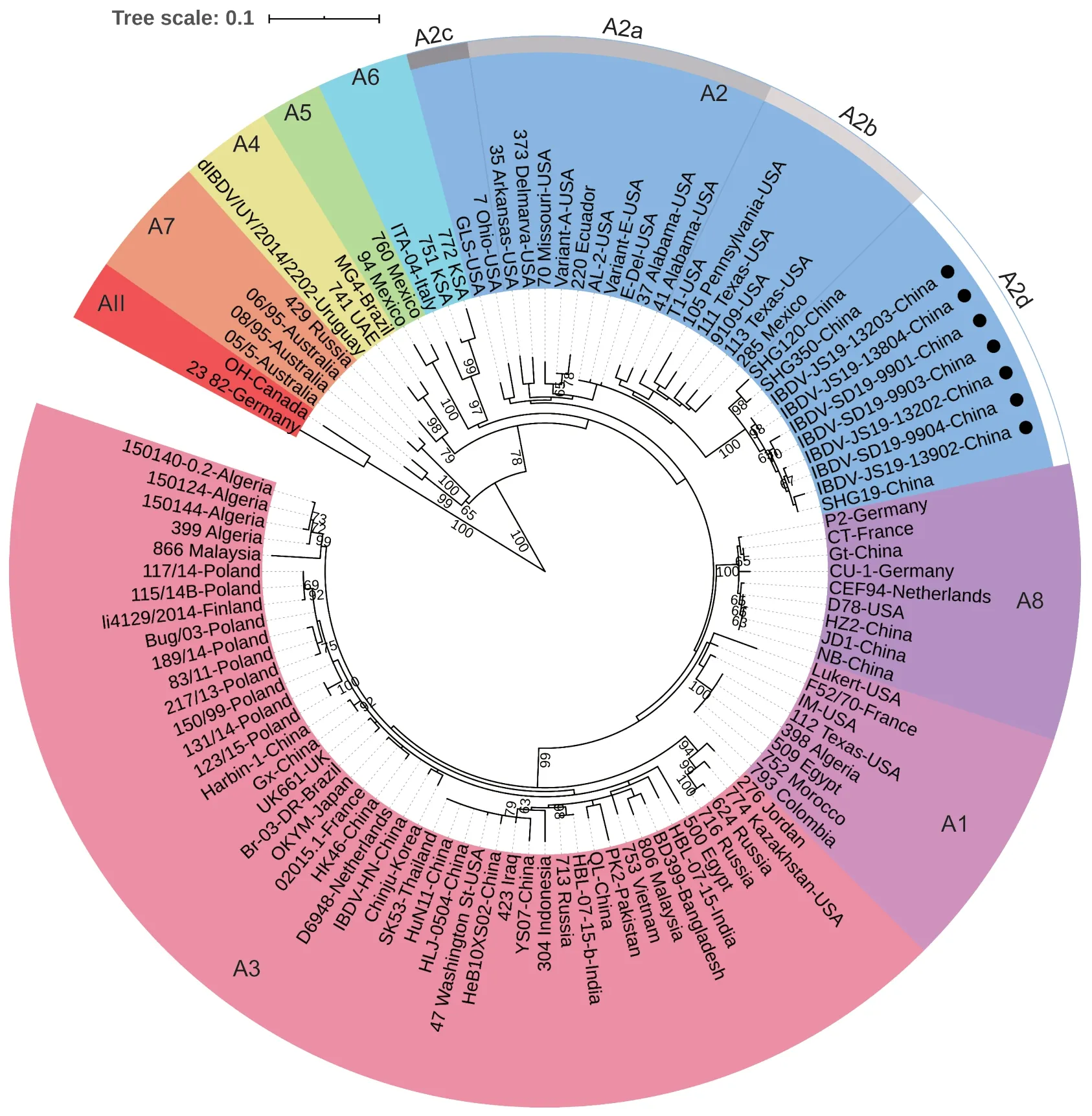
Fig.1 Phylogenetic analysis of the nucleotide sequences of the VP2 hypervariable region (HVR).The tree was generated by the Maximum-likelihood method using MEGA6 Software.The tree is drawn to scale,with branch lengths measured in the number of substitutions per site.Only branches supported by a bootstrap value above 60% are displayed.The analysis involved 105 nucleotide sequences,and the strains detected in this study are highlighted with a solid circle.The nine genogroups based on VP2 HVR of segment A are marked.
Combining the sequence characteristics of VP2 and VP1,an improved scheme for IBDV genotype classification was proposed.In this new scheme,the traditional four phenotypes of IBDV (classic,variant,very virulent,and attenuated IBDV) correspond to genotypes A1B1,A2B1,A3B2,and A8B1,respectively (Table 1).The novel variant strains identified in this study were classified into genotype A2dB1 (Table 1).In addition,five types of recently identified segment-reassortant IBDV strains could be clearly classified as genotypes A3B1 (vv-A/att-B),A3B3 (vv-A/uniq-B),A3BII(vv-A/II-B),A8B2 (att-A/vv-B),and A3B4 (vv-A/trans-B) (vv,very virulent strain;att,attenuated strain;uniq,HLJ0504-like strain;trans,transitional-lineage IBDV;II,serotype II strain)(Appendix C).
4.Discussion
IBDV was first reported in the USA in 1957 (Cosgrove 1962).In 1987,another phenotype of IBDV known as variant strain with different antigenicity was reported,also in the USA,and the initial IBDV strains were designated“classic”strains (Jackwood and Saif 1987).Two years later,a more serious form of IBD with higher mortality,induced by a third phenotype of IBDV,broke out in Europe.Pathogenically,they were designated“very virulent”strains (vvIBDV)(Chettleet al.1989) and the antigenicity of vvIBDV turned out to be similar to the classic strains (Eterradossiet al.1999).In addition,attenuated strains of IBDV developed through blind-passage (Wanget al.2004) or reverse genetics (Gaoet al.2011;Qiet al.2011) are generally used as vaccines.With continuing mutations of IBDV,the traditional phenotypic names are no longer able to encompass all the newly occurring mutated strains (Jackwoodet al.2018).In recentyears,a descriptive scheme has been used to designate new subtypes of IBDV.For example,the similar terms of“distinct”and“distinctive”have been used to describe two obviously different strains isolated in South America (Hernandezet al.2015) and Italy (Lupiniet al.2016),respectively,which has undoubtedly caused some confusion.Since 2017,one newly identified variant IBDV strain associated with severe immunosuppression was designated as novel variant IBDV(Fanet al.2019) to highlight its evolutionarily divergence from the early variant strains identified in the USA.Recently,novel reassortant strains designated transitional-lineage IBDV emerged in Poland and Finland (Tammirantaet al.2018;Pikułaet al.2020).However,the terms“novel variant strains”and“transitional-lineage”are obviously also based on a descriptive naming system (Jackwoodet al.2018).It is inconceivable how to name another novel IBDV with new molecular or antigenic characteristics compared to current IBDV in the future.Therefore,it is essential and pressing to propose a reasonable and sustainable scheme for the classification of IBDV.
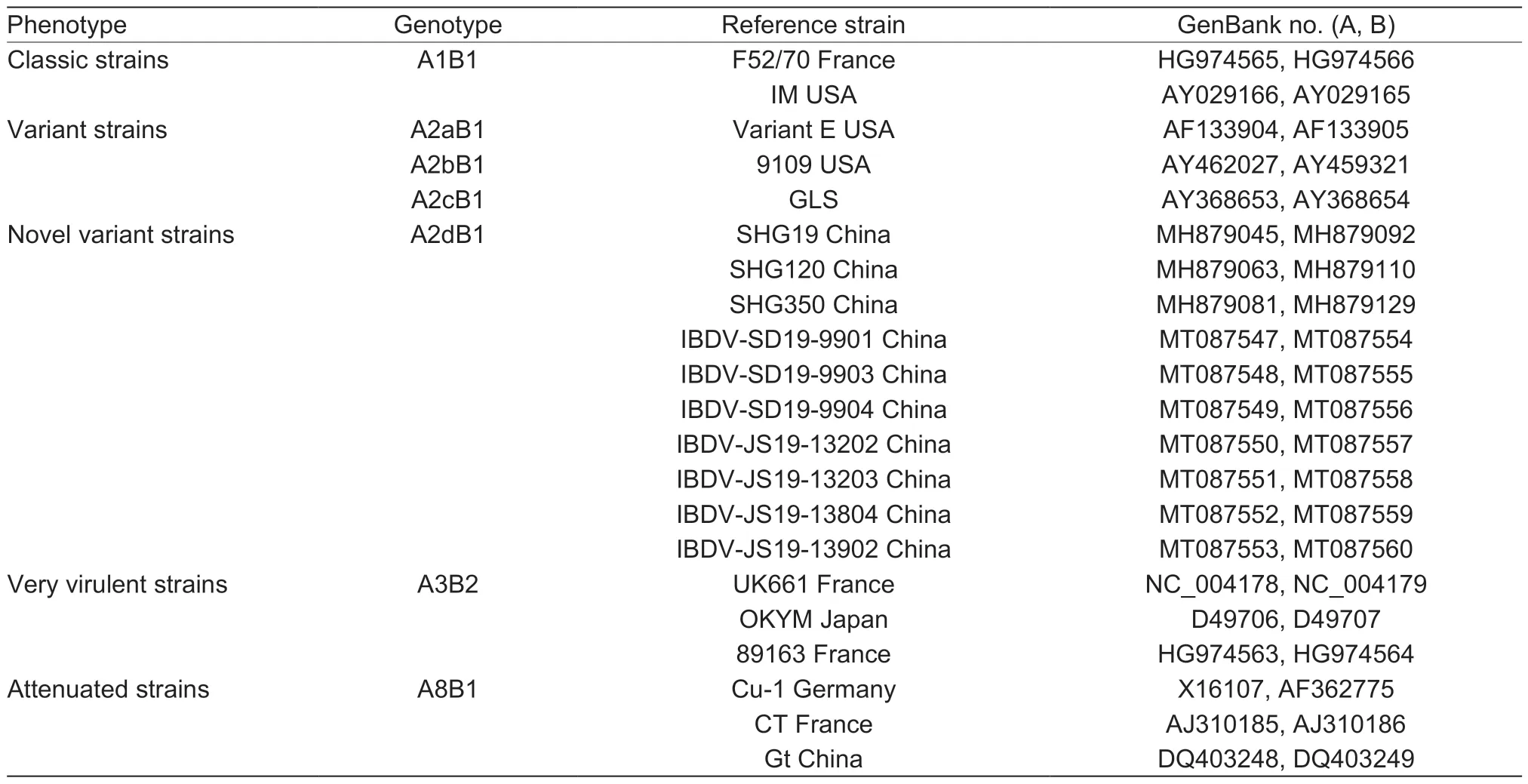
Table 1 The genotype classification corresponding to the traditional phenotypes of infectious bursal disease virus (IBDV)
In 2017,based on the genogroups identified from phylogenetic analysis of the nucleotide sequence of the VP2 HVR,a classification scheme for IBDV was proposed(Michel and Jackwood 2017),which has greatly contributed to the molecular epidemiology of IBDV.According to that new classification scheme,the traditional phenotypes of classic IBDV,variant IBDV,and vvIBDV were classified into genogroups A1,A2,and A3,respectively.In addition,other IBDVs with evolutionary characteristics distinct from the traditional three phenotypes were further classified into genogroups A4,A5,A6,and A7 (Michel and Jackwood 2017).The distinct IBDV (dIBDV) strains isolated in South America (Hernandezet al.2015) formed genogroup A4.Genogroup A5 contained Mexico IBDVs with homologous recombination characteristics of variant and classic strains(Jackwood 2012).The ITA-like strains with a similar identity to the distinctive IBDV identified in Italy (Lupiniet al.2016) were classified as genogroup A6.Some Australian strains (Ignjatovic and Sapats 2002) formed genogroup A7.Furthermore,genogroup A2 was further divided into three lineages of A2a,A2b,and A2c (Jackwoodet al.2018).However,in the VP2 HVR-based evolutionary tree,the novel variant IBDVs,including the strains isolated in this study,were not clustered in any of the above genogroups or lineages,but formed a new cluster named lineage A2d under genogroup A2.In addition,the attenuated strains with special cell-tropism and nonpathogenic characteristics were defined as a new genogroup A8.When combined with genogroup AII of serotype II strains,IBDVs could be classified into a total of nine genogroups based on the VP2 HVR.
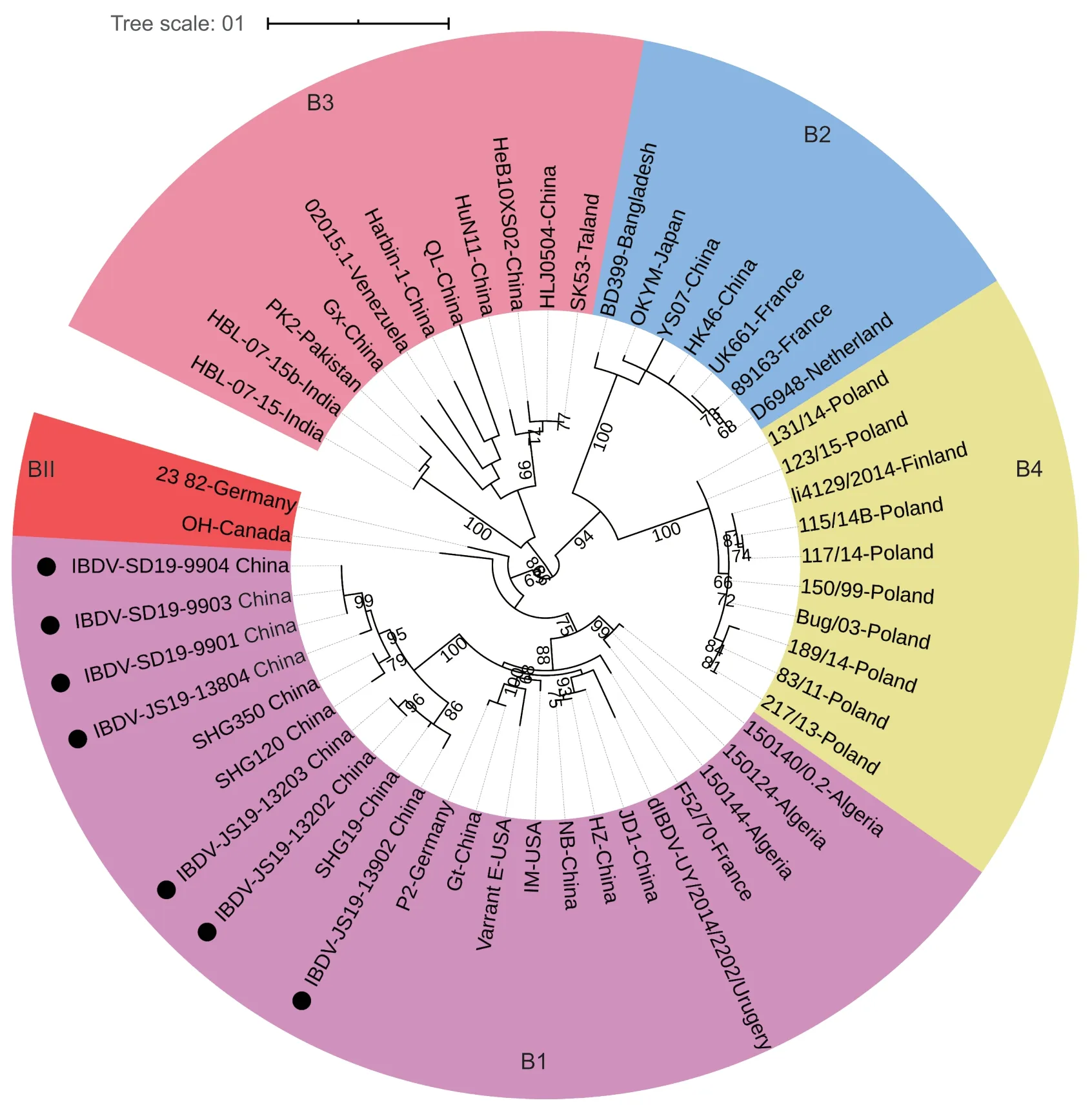
Fig.2 Phylogenetic analysis of the nucleotide sequences of the B-marker of VP1.The tree was generated by the maximumlikelihood method using MEGA6 Software.The tree is drawn to scale,with branch lengths measured in the number of substitutions per site.Only branches supported by a bootstrap value above 60% are displayed.The analysis involved 52 nucleotide sequences,and the strains detected in this study are highlighted with a solid circle.The five genogroups based on VP1 B-marker of segment B are marked.
The genome of IBDV consists of A and B segments.Historically,the evolutionary analyses of IBDV have focused solely on the VP2 gene encoded by segment A,as a number of studies have demonstrated that VP2 is primarily responsible for the virulence,antigenicity,and cell tropism(van den Berg 2000;Brandtet al.2001;Coulibalyet al.2005;Jackwoodet al.2008;Qiet al.2009,2013,2016).Recently,reports have begun to appear that demonstrate a role for segment B,and viral protein VP1 in particular,in the pathogenicity of IBDV (Nouenet al.2012;Escaffreet al.2013).Certain key amino acid residues of VP1 have been shown to be critically involved in the replication and virulence of IBDV (Escaffreet al.2013;Yuet al.2013;Gaoet al.2014).Some studies have even suggested that the VP1 from unknown reservoirs might have played an important role in the evolution and emergence of vvIBDV (Honet al.2006;Alkie and Rautenschlein 2016;Hussainet al.2019).Therefore,it is insufficient for genogroup analyses of IBDV to take only VP2 into consideration;VP1 should also be taken into consideration for the molecular epidemiological study of IBDV.Therefore,we performed a VP1 genogroup analysis.In this study,a phylogenetic tree based on the nucleotide sequences of B-marker of VP1 was constructed.Serotype I strains were clustered into four genogroups (genogroup B1,B2,B3,and B4),while serotype II strains were defined as genogroup BII.VP1 genes have been traditionally divided into two subgroups,vvIBDV (genogroup B2) and non-vv strains (genogroup B1).However,VP1 of HLJ0504-like strains (Qiet al.2011;Heet al.2014;Hussainet al.2019)formed genogroup B3,which may have their origin in other poultry or wild reservoirs (Jackwoodet al.2005;Honet al.2006).HLJ0504-like strains are circulating in China (Qiet al.2011;Heet al.2014),Pakistan (Hussainet al.2019,2020),India (Patelet al.2016),Venezuela (Le Nouenet al.2006),Nigeria (Nwagboet al.2016),and Algeria (Abedet al.2018).Recently,the new group of IBDV strains distinctive from the three canonical groups for VP1 mentioned above was identified in Europe and further classified as transitional group (Tammirantaet al.2018;Pikułaet al.2020).VP1 of European transitional group IBDV formed genogroup B4,for its obvious genetic distance from the IBDV strains in B1,B2,and B3 (Pikułaet al.2020).As a sub-branch with distinct genetic characters,VP1 genes of the novel variant IBDVs isolated in this study were clustered with early variant strains in genogroup B1.
It is more scientific to analyze all relevant genes when performing classification of segmented viruses,such as avian influenza virus.Therefore,we have proposed a new scheme for IBDV genotype classification that simultaneously takes into account both VP2 and VP1.In this new IBDV genotype classification scheme,the four traditional phenotypes of classic IBDV,variant IBDV,vvIBDV,and attenuated IBDV corresponded to genotypes A1B1,A2B1,A3B2,and A8B1,respectively.The novel variant IBDVs identified in this study were classified as genotype A2dB1.
In addition to gene mutations and recombination,genome segment reassortment among various phenotypes has become an important evolutionary feature of IBDV (Le Nouenet al.2006;Hussainet al.2019).To date,five kinds of segment-reassortant strains have been reported,including vv-A/att-B (segment A from very virulent strain and segment B from attenuated strain) (Sunet al.2003;Le Nouenet al.2006;Chenet al.2012;Kasangaet al.2013;Luet al.2015;Nwagboet al.2016;Rajaet al.2016;Leeet al.2017;Pikulaet al.2018),vv-A/uniq-B (segment A from very virulent strain and segment B from HLJ0504-like strain) (Gaoet al.2007;Honet al.2008;Xiaet al.2008;Qiet al.2011;Heet al.2014;Patelet al.2016;Hussainet al.2019),vv-A/trans-B (segment A from very virulent strain and segment B from transitional-lineage strain) (Pikułaet al.2020),att-A/vv-B (segment A from attenuated strain and segment B from very virulent strain) (Weiet al.2008;Cuiet al.2013),and vv-A/II-B (segment A from vvIBDV and segment B from serotype II strain) (Jackwood and Sommer-Wagner 2005;Soubieset al.2017).Genome segment reassortment produced new genotypes,but they had to be named as combined phenotypes,which do not fully reflect their true characteristics.Our new IBDV genotype classification scheme resolves this issue.The descriptive names given to the segment-reassortant IBDVs of vv-A/att-B,vv-A/uniq-B,vv-A/trans-B,att-A/vv-B,and vv-A/II-B can be re-classified as A3B1,A3B3,A3B4,A8B2,and A3BII genotypes in our nomenclature.
To be useful,the new IBDV genotype classification scheme should be flexible enough to accommodate any newly identified genotypes.Compared to VP2,the number of VP1 sequences of IBDV currently available on GenBank is relatively limited.As more information on molecular epidemiology becomes available,this versatile and scalable genotype classification scheme for IBDV will continue to be improved.
5.Conclusion
An improved scheme for IBDV genotype classification was proposed for the first time in this study.Based on the gene characteristics of VP2 (encoded by segment A) and VP1(encoded by segment B),IBDVs were divided into nine“A”genogroups and five“B”genogroups,respectively.The traditional classified phenotypes of classic,variant,very virulent,and attenuated IBDV were shown to correspond to the A1B1,A2B1,A3B2,and A8B1 genotypes in the proposed scheme.The novel variant IBDVs,including the strains identified in this study,were classified as belonging to genotype A2dB1.The flexibility and versatility of this improved classification scheme will allow the unambiguous identification of existing and emerging IBDV strains,which will be of great significance for the molecular epidemiology study of IBDV.
Acknowledgements
This work was supported by the Natural Science Foundation of Heilongjiang Province,China (ZD2020C006 and TD2019C003),the National Key Research and Development Program of China (2016YFE0203200),the Heilongjiang Province Foundation for the National Key Research and Development Program of China (GX18B011),the Major Project of National Natural Science Foundation of China(31430087),and the earmarked fund for China Agriculture Research System (CARS-41-G15).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Ethical approval
All applicable internaltional,national and institutional guidelines for the care and use of animals were followed.
Appendicesassociated with this paper are available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
杂志排行

Journal of Integrative Agriculture的其它文章
- Errata regarding previously published articles
- Ultrastructural studies of seed coat and cotyledon during rapeseed maturation
- Resource utilization of agricultural solid waste
- Can harvest outsourcing services reduce field harvest losses of rice in China?
- The river chief system and agricultural non-point source water pollution control in China
- Exploring the genetic features and signatures of selection in South China indigenous pigs