一个中国全色盲家系CNGB3基因新突变
2021-04-10周钟强彭海鹰史平玲唐贺魏圆梦李苗雷博黄爱国
周钟强 彭海鹰 史平玲 唐贺 魏圆梦 李苗 雷博 黄爱国
河南省人民医院 郑州大学人民医院 河南省立眼科医院 河南省眼科研究所,郑州 450003
全色盲又称典型性全色盲或视杆细胞单色视,是一种罕见的、常染色体隐性遗传视锥细胞功能障碍疾病,主要表现为畏光、眼球震颤、视力下降和色觉异常[1-5]。目前已发现6个全色盲致病基因,分别是环核苷酸门控通道α3(cyclic nucleotide gated channel alpha 3,CNGA3)、环核苷酸门控通道β3(cyclic nucleotide gated channel beta 3,CNGB3)、鸟嘌呤结合蛋白α转导活性肽2(G protein subunit alpha transducin 2,GNAT2)、磷酸二酯酶6C(phosphodiesterase 6C,PDE6C)、磷酸二酯酶6H(phosphodiesterase 6H,PDE6H)和抗体活化转录因子6(activating transcription factor 6,ATF6)[1-6]。CNGA3、CNGB3和GNAT2基因编码的蛋白在视锥细胞的光传导通路中发挥重要作用,PDE6C和PDE6H基因编码视锥细胞环鸟核苷酸(cyclic guanosine monophosphate,cGMP)PDE亚单位,使cGMP转化为5’-GMP,在视锥细胞的视觉传导方面发挥重要作用。ATF6与蛋白正确折叠有着密切关系,其活性减弱会造成内质网氧化应激增加,甚至引起细胞凋亡,与视网膜退行性疾病,包括CNGA3和CNGB3基因缺陷引起的全色盲有着密切关系。这些基因在光传导通路中相互关联,其中任何环节的异常都可能导致全色盲的发生[1-4]。目前已有的全色盲家系研究多为高加索人种,尚缺少中国人群的研究。本研究对1个中国全色盲家系进行遗传学研究,以进行鉴别诊断并确定该家系的致病基因突变。
1 资料与方法
1.1 一般资料
采用家系调查研究方法,对2018年11月在河南省立眼科医院收集的1个来自河南省洛阳市的汉族全色盲家系进行研究。本研究经河南省立眼科医院伦理委员会审核批准[批文号:HNEECKY-2019(15号)],研究过程严格遵循《赫尔辛基宣言》。成年受试者及未成年受试者监护人均了解本研究方法及目的并自愿签署知情同意书。
1.2 方法
1.2.1眼科临床检查 详细询问并记录该家系家族史,进行详细的眼科专科检查。家系所有成员均接受最佳矫正视力(best-corrected visual acuity,BCVA)检查;采用裂隙灯显微镜和前置镜检查患者眼前节和眼底情况;采用阿托品眼用凝胶点眼扩瞳后经电脑自动验光仪验光、检影验光和主觉验光确定受检眼屈光度;采用CR-2 AF型自动免扩瞳眼底照相机(日本佳能公司)进行眼底照相;采用频域光相干断层扫描成像(spectral-domain optical coherence tomography,SD-OCT)仪(RTVue XR 100-2型,美国Optovue公司)检查黄斑区结构;采用孟塞尔色觉测试工具(Munsell FM 100,美国爱色丽公司)进行色觉检查;采用全视野闪光视网膜电图(flash electroretinography,FERG)(RETI-Port 21型眼电生理仪,德国罗兰公司)评估受检眼视网膜功能。由于裂隙灯显微镜和前置镜检查发现Ⅲ1双眼黄斑颞侧、赤道及赤道前视网膜可见斑片状萎缩灶,且赤道部及赤道前视网膜可见明显色素沉着,故补充全景眼底照相仪(Daytona P200T型激光扫描检眼镜,意大利Optos Plc公司)照相。Ⅲ2因无法配合进行FM100孟塞尔色觉检查及FERG检查,改为色盲检查图检查和RETeval便捷式视觉电生理系统(美国LKC公司)检查。
1.2.2DNA提取 采集所有家系成员外周静脉血各4 ml,保存于乙二胺四乙酸处理过的抗凝管中。采用血液基因组DNA提取试剂盒(美国Omega公司),按照标准操作流程提取全基因组DNA,用紫外分光光度计(美国Nanodrop公司)进行定量及质量控制。
1.2.3遗传眼病捕获芯片靶向捕获富集高通量测序 根据中国眼遗传病诊疗小组、中国眼科遗传联盟制定的《眼遗传病基因诊断方法专家共识》[7],首先采用包含441个致病基因的遗传眼病捕获芯片(北京中因科技有限公司)进行靶向捕获富集高通量测序。从先证者(Ⅲ1)及其胞弟(Ⅲ2)、父母外周血中提取基因组DNA,构建全基因组文库,通过Illumina HiSeq高通量测序平台进行DNA测序,二代panel的平均测序深度为500 X,数据量为1 G,对明确的致病性变异采用Sanger测序进行验证。测序仪下机原始数据使用bcl2fastq将.bcl文件转换成.fastq文件,并使用BWA、Samtools和Picard软件将reads比对到人类参考基因组GRCh37/hg19,生成的.bam文件采用GATK系列软件进行局部重新比对和重复序列去除并进行变异检出。采用Annovar对.vcf变异文件进行变异注释。致病变异位点筛选原则:(1)筛选ExAC、1000 Genomes、gnomAD等数据库中未见正常人携带或携带频率小于5%的变异。(2)使用SIFT、Polyphen2、MutationTaster等多种蛋白功能预测软件进行基因变异位点分析;采用dbscSNV_ADA_SCORE和dbscSNV_RF_SCORE预测剪切位点变异。(3)参考OMIM、HGMD、ClinVar等多种数据库对变异位点进行致病性评估,对明确的致病性变异采用Sanger测序进行验证。
1.2.4人全基因组测序 由于应用包含441个致病基因的遗传眼病捕获芯片进行靶向捕获富集高通量测序未获得有意义的致病基因及突变位点,故对Ⅲ1及其父母、Ⅲ2共4人的DNA进行全基因组测序(whole-genome sequencing,WGS)。从受检者外周血中提取基因组DNA,构建全基因组文库,通过Illumina HiSeq高通量测序平台测序,平均测序深度为30 X,数据量为90 G。对明确的致病性变异采用Sanger测序进行验证。测序仪下机原始数据使用bcl2fastq将.bcl文件转换成.fastq文件,并使用BWA、Samtools和Picard软件将reads比对到人类参考基因组GRCh37/hg19,生成的.bam文件采用GATK系列软件进行局部重新比对,重复序列去除并进行变异检出。使用Annovar对.vcf变异文件进行变异注释。致病变异位点筛选原则:(1)筛选ExAC、1000 Genomes、gnomAD等数据库中未见正常人携带或携带频率小于5%的变异。(2)使用SIFT、Polyphen2、MutationTaster等多种蛋白功能预测软件进行基因变异位点分析。(3)参考OMIM、HGMD、ClinVar等多种数据库对变异位点进行致病性评估。
1.2.5生物信息学分析 对除Ⅲ1以外的其他家系成员进行Sanger测序,验证WGS确定可疑致病基因及突变位点,并对发现的可疑基因变异在HGMD中查看是否为已报道致病突变,在1000 Genome(http://browser.1000genomes.org/index.html)、EVS(http://evs.gs.washington.edu/EVS/)和ExAC(http://exac.broadinstitute.org/)数据库中查看变异在人群中的基因频率;采用Polyphen-2和SIFT软件预测c.1285dupT对蛋白功能的影响;采用dbscSNV和SPIDEX软件预测分析c.129+1G>A对蛋白质翻译的影响;结合家系图与先证者进行共分离分析。
2 结果
2.1 全色盲家系成员临床特征
该家系共3代10位成员,其中全色盲患者2例,分别为Ⅲ1及Ⅲ2,其他家系成员表型均正常,符合常染色体隐性遗传(图1)。Ⅲ1,女,14岁,视力右眼-4.25/-2.75×180→0.3,左眼-3.50/-3.50×175→0.3;Ⅲ2,男,5岁,视力右眼+3.75/-1.75×30→0.2+,左眼+1.25/-1.75×160→0.3-。2例患儿均自幼视力低下、畏光,随着年龄增长症状无明显变化。患者均为中心凹注视,未见眼球震颤,眼球运动正常,双眼眼前节正常,玻璃体透明。Ⅲ1双眼视盘色稍淡,视网膜呈豹纹状,动脉稍细,黄斑颞侧、赤道及赤道前视网膜可见斑片状萎缩灶,赤道及赤道前视网膜可见明显色素沉着,黄斑中心凹反光可见;SD-OCT检查显示双眼黄斑区外界膜及椭圆体带反射信号欠规则;FERG检查示双眼暗视0.01刺激ERG b波、暗视3.0和暗视10.0刺激下ERG a、b波振幅均轻度下降,明视3.0刺激下ERG a、b波振幅严重下降,30 Hz ERG各波记录不到;FM100孟塞尔色觉检查示双眼为全色盲。Ⅲ2双眼视盘色泽尚可,视网膜动、静脉及后极部视网膜未见明显异常,赤道及赤道前视网膜可见明显色素沉着,黄斑中心凹反光可见;SD-OCT、FERG检查结果与Ⅲ1表现一致,色盲检查图检查提示为全色盲(图2~5)。
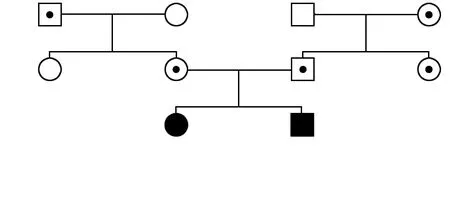
图1 先证者家系图 CNGB3基因突变位点c.129+1G>A和CNGB3 c.1285dupT在该家系中患儿、携带者和正常表型成员之间共分离 □:正常男性;○:正常女性;·:男性携带者;⊙:女性携带者;■:男性患者;●:女性患者;:先证者Figure 1 Pedigree of the family with achromatopsia The compound heterozygous mutations,c.129+1G>A and c.1285dupT,in CNGB3 were segregated among patients,carriers and unaffected family members □:normal male;○:normal female;·:male carrier;⊙:female carrier;■:male patient;●:female patient;:proband
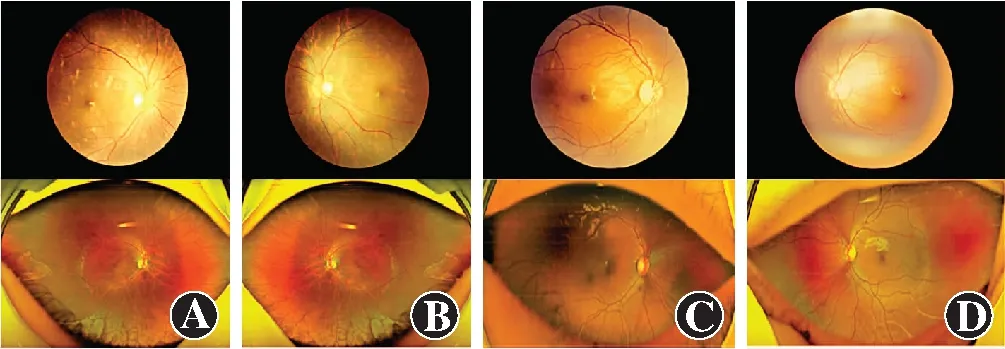
图2 Ⅲ1和Ⅲ2双眼免扩瞳眼底照相及全景眼底照相 Ⅲ1双眼视盘色稍淡,视网膜动脉稍细,后极部视网膜呈豹纹状;可见后极部、赤道及赤道前斑片状萎缩灶及色素沉着。Ⅲ2双眼视盘色泽尚可,视网膜动静脉未见明显改变;赤道及赤道前视网膜可见明显色素沉着 A:Ⅲ1右眼眼底彩色照相和全景眼底照相 B:Ⅲ1左眼眼底彩色照相和全景眼底照相 C:Ⅲ2右眼眼底彩色照相和全景眼底照相 D:Ⅲ2左眼眼底彩色照相和全景眼底照相Figure 2 Fundus color photography and ultra widefield fundus color photography of Ⅲ1 and Ⅲ2 Fundus color photography of both eyes of Ⅲ1 showed that optic nerve heads were pale,and retinal arteries were thinned,and the retina of the posterior pole turned into leopard-like.Ultra widefield fundus color photography of Ⅲ1 displayed patchy atrophy in the temporal lateral retina of macula and pigmentary mottling at the posterior pole and the equator and in the preequatorial retina.Fundus color photography of both eyes of Ⅲ2 showed normal fundus in both eyes,and pigmentary mottling could be seen at the equator and in the preequatorial retina A:Images of the right eye of Ⅲ1 B:Images of the left eye of Ⅲ1 C:Images of the right eye of Ⅲ2 D:Images of the left eye of Ⅲ2
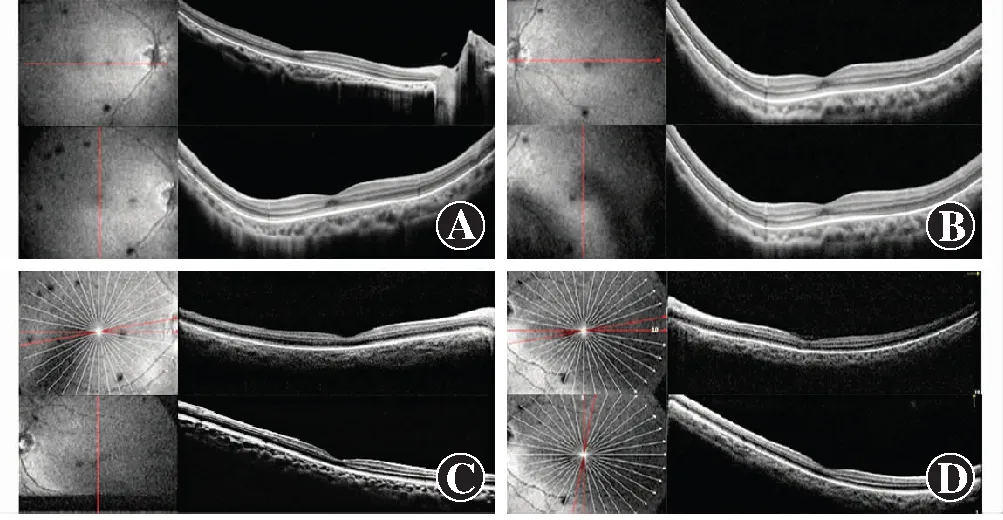
图3 Ⅲ1和Ⅲ2双眼黄斑区SD-OCT检查 Ⅲ1和Ⅲ2双眼黄斑区外界膜及椭圆体带反射信号欠规则 A:Ⅲ1右眼 B:Ⅲ1左眼 C:Ⅲ2右眼 D:Ⅲ2左眼Figure 3 The SD-OCT findings in macula of Ⅲ1 and Ⅲ2 Reflection bands of external membrane and ellipsoid line were irregular in the two patients A:The right eye of Ⅲ1 B:The left eye of Ⅲ1 C:The right eye of Ⅲ2 D:The left eye of Ⅲ2
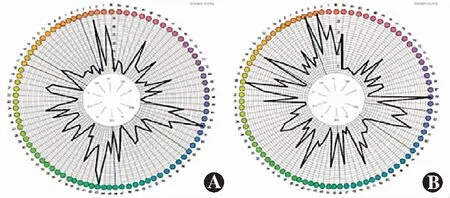
图4 Ⅲ1双眼FM100孟塞尔色觉检查 右眼检查总错误得分为776,左眼为880,双眼均不能分辨红、绿和蓝色 A:右眼 B:左眼Figure 4 Farnsworth Munsell 100 hue test results of both eyes of Ⅲ1 The total error scores of her right and left eyes were 776 and 880,respectively,which meant that the eyes could not discriminate red,green and blue A:Right eye B:Left eye
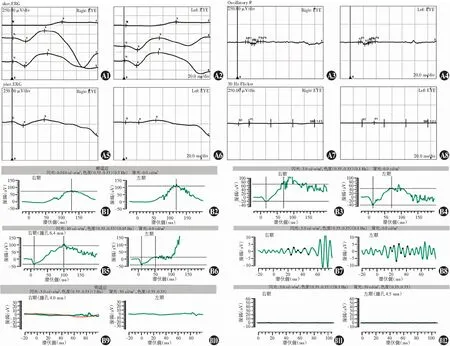
图5 Ⅲ1和Ⅲ2双眼FERG表现 A:Ⅲ1双眼FERG波形 A1为右眼暗视0.01、3.0和10.0 ERG波形;A2为左眼暗视0.01、3.0和10.0 ERG波形;A3为右眼暗视3.0振荡电位;A4为左眼暗视3.0振荡电位;A5为右眼明视3.0 ERG波形;A6为左眼明视3.0 ERG波形;A7为右眼明视3.0闪烁光ERG;A8为左眼明视3.0闪烁光ERG B:患者Ⅲ2双眼FERG B1为右眼暗视0.01 ERG;B2为左眼暗视0.01 ERG;B3为右眼暗视3.0 ERG;B4为左眼暗视3.0 ERG;B5为右眼暗视10.0 ERG;B6为左眼暗视10.0 ERG;B7为右眼暗视3.0闪烁光ERG;B8为左眼暗视3.0闪烁光ERG;B9为右眼明视3.0 ERG;B10为左眼明视3.0 ERG;B11为右眼明视3.0闪烁光ERG;B12为左眼明视3.0闪烁光ERGFigure 5 The FERG findings of Ⅲ1 and Ⅲ2 A:FERG findings of Ⅲ1 A1 was a-and b-waves of scotopic 0.01,3.0 and 10.0 ERG of the right eye;A2 was a-and b-waves of scotopic 0.01,3.0 and 10.0 ERG of the left eye;A3 was wavelets of scotopic 3.0 oscillatory potentials of the right eye;A4 was wavelets of scotopic 3.0 oscillatory potentials of the left eye;A5 was a-and b-waves of photopic 3.0 ERG of the right eye;A6 was a-and b-waves of photopic 3.0 ERG of the left eye;A7 was wavelets of photopic 30Hz ERG of the right eye;and A8 was wavelets of photopic 30Hz ERG of the left eye B:FERG findings of Ⅲ2 B1 was a-and b-waves of scotopic 0.01 ERG of the right eye;B2 was a-and b-waves of scotopic 0.01 ERG of the left eye;B3 was a-and b-waves of scotopic 3.0 ERG of the right eye;B4 was a-and b-waves of scotopic 3.0 ERG of the left eye;B5 was a-and b-waves of scotopic 10.0 ERG of the right eye;B6 was the a-and b-waves of scotopic 10.0 ERG of the left eye;B7 was wavelets of scotopic 30 HZ ERG of the right eye;B8 was wavelets of scotopic 30 HZ ERG of the right eye;B9 was a- and b-waves of photopic 3.0 ERG of the right eye;B10 was a-and b-waves of photopic 3.0 ERG of the left eye;B11 was wavelets photopic 30Hz ERG of the right eye;and B12 was wavelets photopic 30Hz ERG of the left eye
2.2 全色盲家系CNGB3基因检查
WGS测序发现,Ⅲ1和Ⅲ2在CNGB3基因上存在c.129+1G>A和c.1285dupT的复合杂合突变。Sanger测序验证证实Ⅲ1和Ⅲ2存在CNGB3基因c.129+1G>A和c.1285dupT复合杂合突变(图6)。
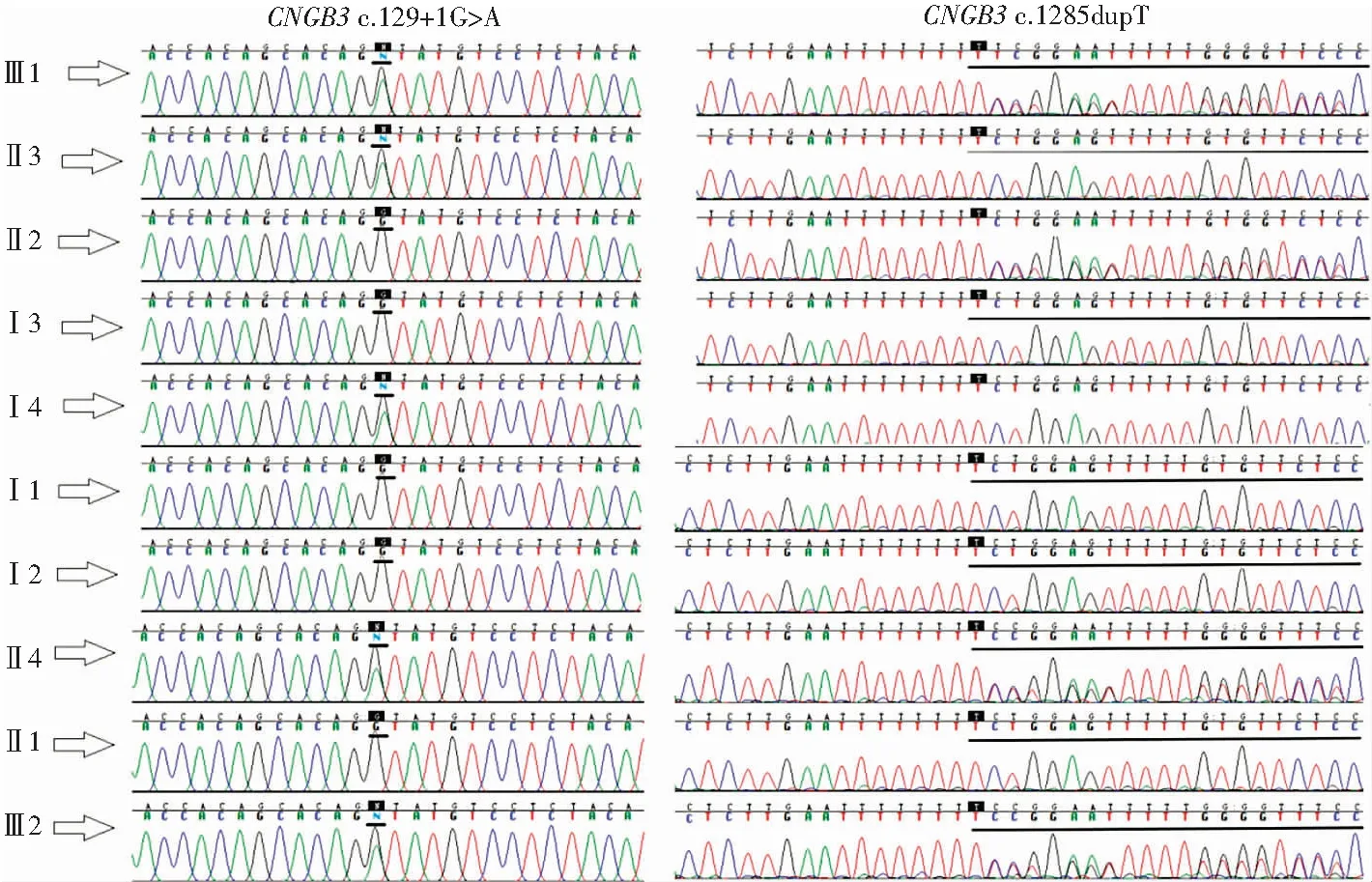
图6 CNGB3基因突变位点 CNGB3基因在全色盲家系Ⅲ1及Ⅲ2存在c.129+1G>A和c.1285dupT复合杂合突变,家系中表型正常成员Ⅰ4、Ⅱ3和Ⅱ4仅存在杂合突变c.129+1G>A,Ⅰ1和Ⅱ2仅存在杂合突变c.1285dupTFigure 6 Mutations of CNGB3 gene In this pedigree,both Ⅲ1 and Ⅲ2 carried both CNGB3 c.129+1G>A and CNGB3 c.1285dupT,whileⅠ4,Ⅱ3 and Ⅱ4 carried CNGB3 c.129+1G>A,andⅠ1 and Ⅱ2 carried CNGB3 c.1285dupT
2.3 基因突变致病性和共分离分析
经过生物信息学软件分析CNGB3基因c.129+1G>A和c.1285dupT均为致病突变。CNGB3的突变c.129+1G>A在1号外显子后的内含子的第1个碱基,其在千人汉族人群及千人东亚人群中的突变频率为0。CNGB3突变c.1285dupT在11号外显子上,其在千人汉族人群及千人东亚人群中的突变频率为0。
DNA样本测序表明,CNGB3基因的杂合突变c.129+1G>A遗传自祖母Ⅰ4,杂合突变c.1285dupT遗传自外祖父Ⅰ2。家系中表型正常者Ⅰ4、Ⅱ3和Ⅱ4仅携带杂合突变c.129+1G>A,Ⅰ1和Ⅱ2仅携带杂合突变c.1285dupT。家系成员Ⅰ2、Ⅰ3和Ⅱ1中2个突变都不存在。该家系符合常染色体隐性遗传规律和家系共分离(图1)。
3 讨论
目前已发现全色盲的致病基因有6个,其中CNGB3基因为最常见的致病基因[8]。迄止目前已经发现,全色盲患者CNGB3基因突变的致病方式至少包括114种复合杂合突变、32种纯合突变、1种杂合突变及其他方式[5,8-11]。本家系中Ⅲ1和Ⅲ2均存在CNGB3基因c.129+1G>A和c.1285dupT复合杂合突变,根据《ACMG遗传变异分类标准中文版专家共识指南》,该复合杂合突变为该全色盲家系的致病突变,且本研究发现的CNGB3基因c.129+1G>A和c.1285dupT复合杂合突变不同于已报道的114种复合杂合突变,为新发致病突变。
CNGB3基因定位于8q21-22,由18个外显子组成,编码809个氨基酸[1]。视锥细胞CNG阳离子通道位于光级联传导通路的末端,直接被cGMP激活,控制光感受器细胞外节质膜的离子流,在光激发红、绿、蓝敏感的视锥细胞产生的生物电反应方面发挥重要作用[1],该通道的β亚单位即为CNGB3基因编码的蛋白之一,CNGB3基因的多种突变均可导致视锥细胞的光传导通路异常,导致全色盲的发生[1]。本研究发现的CNGB3基因剪切位点杂合突变c.129+1G>A使第1个内含子无法正常剪切,从而导致蛋白翻译提前终止,使视锥细胞CNG阳离子通道的β亚单位不能正常合成,最终对视锥细胞的光传导通路产生影响,这种影响会像其他突变一样导致全色盲的发生。虽然CNGB3基因c.129+1G>A纯合突变会导致视锥、视杆细胞营养不良[12],但同源染色体中仅存在1个该突变,是否会导致全色盲以及通过何种机制导致全色盲还有待进一步研究。本研究发现的另1个突变c.1285dupT在编码区的第1 285位处多了1个核苷酸T,造成移码突变,蛋白翻译提前终止,最终产生1个只有460个氨基酸的错误蛋白产物。虽然在Mayer等[11]的研究中该突变为其中3个家系的致病突变,并且无论是CNGB3基因c.1285dupT纯合突变还是c.1148delC和c.1285dupT复合杂合突变均可致病,但仅1条同源染色体携带致病基因而另1条同源染色体为野生型是否会导致全色盲,以及致病基因导致全色盲的机制还有待进一步研究。
由于本研究家系中的致病突变携带者无临床表现,因此我们推断CNGB3基因c.129+1G>A或c.1285dupT分别与野生型杂合并不具备致病性,因为携带者还有1个野生型等位基因可以用来合成正常蛋白。当CNGB3基因同时存在c.129+1G>A和c.1285dupT突变,2个等位基因都无法产生功能正常的蛋白时即会致病。
虽然Ⅲ1和Ⅲ2全色盲的诊断明确,但要对其进行分类非常困难。根据既往研究,全色盲分为完全型和不完全型[1-4]。部分完全型全色盲患者会在视网膜中周部出现视网膜色素上皮异常,可表现为色素增生或萎缩。全色盲患者FERG显示视锥细胞反应记录不到而视杆细胞反应基本正常[1-4]。本家系的患者表现为视锥系统功能严重下降,而视杆系统功能轻度下降,因此本研究认为虽然2例患儿ERG的表现与文献报道的ERG表现并不完全相符[1-4],但仍符合全色盲的ERG表现。Ⅲ1和Ⅲ2的双眼BCVA分别为0.3和>0.2,除眼底赤道部及赤道前视网膜有色素沉着,Ⅲ1还存在黄斑颞侧、赤道及赤道前视网膜斑片状萎缩灶,导致很难对其进行分类,既往研究发现完全型全色盲BCVA一般低于0.1[1-4],中周部视网膜色素上皮异常往往出现在完全型全色盲者,而本家系患者视力和视网膜色素沉着部位均不能归为完全型全色盲,其机制仍待进一步研究。
本家系2例患儿因视力低下和畏光就诊,眼部检查均提示其视锥系统功能异常,虽然色盲图检查Ⅲ1为色盲,但就诊时未能确定究竟是视锥细胞营养不良还是全色盲。为了进一步进行鉴别诊断,本研究首先采用包含441个致病基因的遗传眼病捕获芯片进行靶向捕获富集高通量测序以进行基因诊断,虽然选取的panel包含所有已知的眼部遗传疾病的致病基因,但未能获得本家系的有意义的致病基因。为找到致病突变,本研究进一步通过WGS确定了致病突变。
本研究发现了全色盲患者1个新的突变位点及1种新的致病方式,补充了全色盲的基因突变谱。但本研究仍存在以下局限性:(1)仅调查了1个家系,研究的外推性欠佳;(2)本研究既采用了免扩瞳眼底照相,也采用了全景眼底照相,但在实际工作中免扩瞳眼底照相加拼图可以替代全景眼底照相;(3)本家系在进行基因检测时未先采用仅针对眼底遗传性疾病的靶向基因测序,而直接采用了包含441个致病基因的遗传眼病捕获芯片进行靶向捕获富集高通量测序,且在测序未得到理想结果时又未先进行全基因外显子组测序,而直接进行了WGS,这未能很好地遵循眼遗传病基因检测流程,如果本研究最初选取的是更精准的眼底遗传疾病的panel,或先对有疑问的位点单独设计引物进行测序,也许实验过程会更优化、更经济,也更符合遗传疾病研究的最优诊断路径,不仅能增强检测的实效性,还会降低检测费用。
虽然本研究存在一些不足,但是本研究发现了全色盲CNGB3基因1个新的致病突变位点及1个新的复合杂合突变方式,扩大了CNGB3基因的突变谱,对更深入地了解全色盲的发病机制提供了有价值的信息,同时也为将来全色盲的靶向基因治疗及患者家族成员的优生优育选择提供了一定的依据。
利益冲突所有作者均声明不存在利益冲突
