Fe物种改性海胆状Nb2O5光催化降解类吩噻嗪染料
2021-04-10牛浩通马瑞婧王会香吕宝亮
畅 通 牛浩通 马瑞婧 王会香 吕宝亮
(1运城学院应用化学系,运城 044000)
(2中国科学院山西煤炭化学研究所煤转化国家重点实验室,太原 030001)
(3太原理工大学材料科学与工程学院,太原 030024)
(4运城学院物理与电子工程系,运城 044000)
0 引 言
现代印染工业的快速发展带来了严重的有机染料水污染问题。类吩噻嗪染料是一类用量大但难于后处理的有机染料,其在水中的长时间存在严重威胁着人类及生物的健康,因此,如何有效去除这类染料是污水处理的一个重要研究课题[1-2]。光催化技术具有反应条件温和、反应设备简单、二次污染小、易于操作控制等优点。在污水治理领域,光催化降解技术可以将许多难降解的有机物转化为H2O、CO2等无污染小分子,达到完全无机化的目的[3]。然而,目前的半导体光催化剂普遍存在因光生载流子复合率高而导致降解效率低的问题[4-5]。因此,开发新的高效催化剂和提高现有材料的光催化效率成为众多研究者的目标。
Nb2O5是一种重要的n型宽禁带(Eg)半导体材料,其室温下Eg为3.1~4.0 eV,紫外区的光吸收类似于TiO2,且化学性质稳定,因此被认为是具有开发前景的光催化半导体材料[6-9]。众所周知,催化剂的结构影响其性能,大比表面可以暴露更多的原子,从而提供充足的反应活性位点,因此,制备具有大比表面的Nb2O5是研究人员努力的方向之一。三维分级结构具有中心且向三维方向生长的特性,从而使得催化剂的比表面积大于特定维度的催化剂比表面积,其中海胆状是一种典型的三维分级结构,比如海胆状 WO3、Co3O4、TiO2和 MnO2等[10-14]。
最近,有学者用水热法成功制备出了海胆状Nb2O5纳米球,并将其用于光催化降解有机污染物,表现出了良好的降解活性[15]。然而,光生载流子的高复合率仍是阻碍单一海胆状Nb2O5纳米球光催化降解效率提高的主要原因。为了进一步提高Nb2O5光催化降解活性,需要通过合理的手段对其表面进行修饰。表面半导体复合的方法是降低催化剂催化过程中光生载流子复合的有效途径之一,且已广泛用于提高光催化剂活性的研究中[5,16-17]。因此,我们设计了在Nb2O5纳米球表面一步法原位复合Fe物种的策略,以期改善海胆状Nb2O5纳米球的光催化活性。原因有二:(1)Fe的氧化物和含氧酸盐一般均为半导体,且具有较低的导带位置,可以与Nb2O5能带匹配,有望分离Nb2O5导带上的光生电子,使得其价带上保留的空穴可以对有机污染物进行氧化;(2)在光催化降解过程中加入少量的H2O2,使其与Fe发生 Fenton反应(Fe2++H2O2→ Fe3++OH-+·OH,Fe3++e-→Fe2+),此过程可以快速消耗掉光生电子,进一步抑制光生电子和空穴的复合,并产生大量的羟基自由基用于降解有机污染物[18-20]。
我们在合成海胆状Nb2O5纳米球的基础上,向体系中直接引入Fe3+,通过一步水热法成功制备了Fe物种修饰的海胆状Nb2O5纳米球。与单一海胆状Nb2O5纳米球相比,制备的复合催化剂在全光照射下表现出了良好的光催化活性,能够快速发生Fenton反应,高效且选择性地降解类吩噻嗪亚甲基蓝(MB)和甲苯胺蓝(TB)染料,催化剂易于回收再利用。最后,对光催化降解机理做了合理的阐述。
1 实验部分
1.1 催化剂制备
样品制备过程如下:取2 mmol Nb(HC2O4)5·xH2O(安耐吉)和 0.2 mmol Fe(NO3)3·9H2O(国药)溶于 20 mL去离子水中,超声溶解至溶液完全澄清,然后向溶液中加入80 mL乙醇,继续超声10 min后将以上溶液倒入150 mL自压反应釜,密封后于200℃反应12 h,待温度降到室温后打开反应釜,离心分离固体样品,用去离子水和无水乙醇洗涤干净,60℃烘干6 h,最后将样品置于马弗炉中500℃高温煅烧2 h,即得目标产物,标记为NbFex,x为Fe和Nb的物质的量之比(nFe/nNb),x=0、0.05、0.10和0.20。
1.2 表 征
样品的晶型由粉末X射线衍射仪(XRD,D8 Advance)测定,管电压40 kV,管电流100 mA,辐射源为 Cu Kα 靶(λ=0.154 06 nm),扫描范围为 10°~70°,步长为0.06°;样品微观形貌通过场发射扫描电子显微镜(SEM,JSM-7001F)观察,工作电压10 kV,电流0.1 nA;样品晶格参数由透射电子显微镜(TEM,JEM-2100F)观察,工作电压200 kV,电流200µA;催化剂表面化学组成及价态采用X射线光电子能谱仪(XPS,ESCALAB 250XI)检测,仪器辐射源为Al Kα靶。采用Brunauer-Emmett-Teller(BET)方程和Barrett-Joyner-Halenda(BJH)模型计算N2吸附-脱附测试(MicromeriticsASAP3000)中样品的比表面积以及孔径分布等参数,在通入N2前,200℃抽真空12 h;采用光致发光谱(PL,F-7000)检测半导体光生电子和空穴的复合程度。催化剂的傅里叶变换红外光谱(FT-IR,Nicolet iS50)测试采用 KBr压片,范围400~4 000 cm-1。样品的紫外-可见光(UV-Vis)吸收谱由紫外分光光度计UV-3150测试得到。
1.3 光催化测试
光催化反应器为带冷却水套的100 mL石英杯(横截面积28 cm2),光源为北京泊菲莱科技有限公司生产的PLS-SXE300UV氙灯光源,全光谱波长输出范围为320~780 nm,光源和石英杯之间的垂直距离是15 cm。具体光催化反应如下:取10 mg催化剂分散于50 mL 20 mg·L-1的染料溶液中,在黑暗中超声分散5 min后,在温度10℃的条件下黑暗中搅拌30 min,使染料达到吸附-脱附平衡,随后取样3 mL计算吸附量,然后加入0.1 mL H2O2(质量分数30%),搅拌几秒后再取3 mL样品,浓度记为c0,打开光源,间隔一定时间取样3.0 mL,以7 500 r·min-1的转速离心分离催化剂后用紫外分光光度计测定染料的吸光度。MB、TB、罗丹明B(RhB)的吸收波长分别位于666、634、554 nm,光催化降解效率(R)通过以下公式计算得到:

其中c是反应时间t时的染料浓度,c0是开启光源反应前(t=0)的染料浓度,A和A0分别是对应的吸光度值。
2 结果与讨论
2.1 样品的结构及形貌分析
NbFex(x=0、0.05、0.10、0.20)样品的XRD图如图1所示。NbFe0样品的衍射峰均对应于六方Nb2O5(h-Nb2O5,PDF No.028-0317),其(001)晶面所对应衍射峰的相对强度明显强于其余晶面所对应的衍射峰,且不同于标准卡片的峰强度,说明样品的(001)晶面具有明显的择优生长取向[15]。引入Fe3+后,Nb2O5的(001)晶面衍射峰略微发生右移,这可能是水热合成过程中少量Fe3+进入Nb2O5的晶格所致,因为Fe3+的离子半径小于Nb5+的离子半径(兰氏化学手册中Fe3+:0.064 nm;Nb5+:0.070 nm),部分晶格取代在合成过程中很难避免[21]。此外,引入Fe3+后样品的晶型没有发生变化,然而衍射峰强度减弱,且随着Fe含量的增加,衍射峰强度减弱更明显,说明Fe3+的引入阻碍了Nb2O5的结晶。NbFe0.10和NbFe0.20样品在 33.2°、35.6°和 54.1°处逐渐出现了低结晶度 α-Fe2O3(PDF No.033-0664)的特征衍射峰,分别对应(104)、(110)和(116)晶面。除α-Fe2O3外,3个含Fe样品的XRD图中均没有发现其他Fe物种的衍射峰。
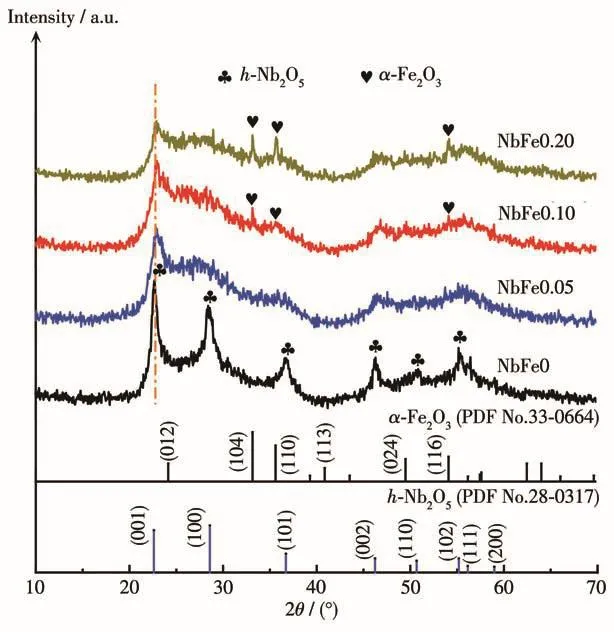
图1 NbFex(x=0、0.05、0.10、0.20)样品的XRD图Fig.1 XRD patterns of NbFex samples(x=0,0.05,0.10,0.20)
采用FT-IR对NbFex样品进一步进行结构分析(图2)。对于NbFe0样品,3 433和1 633 cm-1处出现的峰分别对应于表面吸附水分子中—OH的伸缩振动和弯曲振动;1 400 cm-1处的峰归属于表面羟基的弯曲振动;670 cm-1处的宽峰对应于O—Nb—O多面体的对称性伸缩振动;895 cm-1处的吸收峰对应于表面Nb=O的伸缩振动[22-24]。对于NbFex(x=0.05、0.10、0.20)样品,其FT-IR谱图与NbFe0相似,说明Fe3+的引入虽然对Nb2O5的(001)晶面间距稍有影响(图1),但对其整个晶格结构影响不大,即少量Fe3+引入之后产物主体依然是Nb2O5。此外,红外谱图中没有明显的Fe物种的红外特征,主要是因为O—Fe—O键的振动同样在500~800 cm-1之间[25],且含量不高,所以4个样品表现出了近似相同的红外谱图。
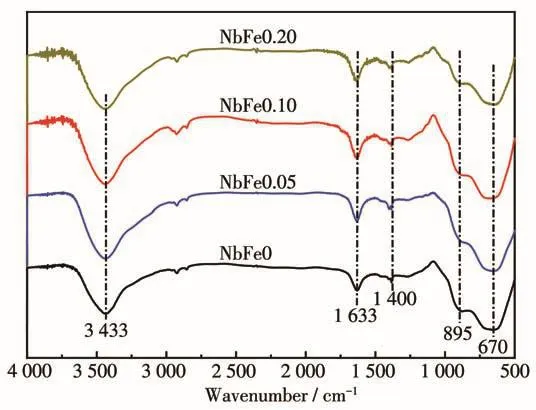
图2 NbFex(x=0、0.05、0.10、0.20)样品的FT-IR谱图Fig.2 FT-IR spectra of NbFex(x=0,0.05,0.10,0.20)samples
样品NbFe0的SEM照片如图3a、3b所示,可以看出,没有引入Fe3+时,实验制备的纯Nb2O5具有明显的海胆状结构,直径在250~400 nm,分散度较好,表面有明显的均匀分布的纳米棒。当引入少量的Fe3+(x=0.05,图3c、3d)后,样品形貌和分散情况均无太大变化,但是样品尺寸略有减小;当x=0.10(图3e、3f)时,样品直径显著减小,发生团聚现象,表面纳米棒长度变短;当x=0.20(图3g、3h)时,纳米球直径增大,表面纳米棒变得不明显。由SEM图可知,Fe3+的引入量较少(x<0.20)时阻碍了Nb2O5中心纳米球和表面纳米棒的生长。当x=0.20时,由于Fe物种的自生长加速(图1),Fe3+对Nb2O5中心纳米球的生长抑制作用减弱,然而表面形成的Fe物种同样减弱了表面纳米棒的生长。总之,引入Fe3+后依然可以得到海胆状结构产物Nb2O5。

图3 NbFex样品的SEM图Fig.3 SEM images of NbFex samples
用TEM和高分辨透射电镜(HRTEM)对NbFe0和NbFe0.10(本实验中光催化效率最高)样品的微观结构进一步进行观察。由NbFe0样品的TEM图(图4a、4b)可知,该样品由直径分布在250~400 nm的海胆状纳米球组成。表面纳米棒的HRTEM(图4c)显示其晶格条纹垂直于纳米棒轴线方向,间距是0.392 nm,对应于h-Nb2O5的(001)晶面,说明纳米棒沿[001]方向生长,可以用定向附着机制解释纳米棒的生长[15,22]。对于NbFe0.10,海胆状形貌没有改变,纳米球的直径分布更为均匀,约为250 nm(图4d),垂直表面分布的纳米棒长度缩短(图4e),结果和SEM一致。图4f是NbFe0.10样品表面纳米棒的HRTEM图,可以看到其晶格条纹间距是0.384 nm,略有减小,对应于XRD中(001)晶面的略微右移。此外,在HRTEM图中没有发现明显的大颗粒Fe物种和其对应的晶格条纹,而能量散射X射线谱(EDS)分析(图4g、4h)和元素映射图(图 4i~4k)则显示 Fe元素的存在,且均匀分布于样品表面,说明制备过程中形成的Fe物种可能以低结晶度或者很小的纳米颗粒存在于Nb2O5纳米球的表面。结合XRD和FT-IR分析结果,进一步说明引入Fe3+对海胆状Nb2O5的微观形貌和晶体结构均无明显影响,形成的Fe物种主要均匀复合在Nb2O5纳米球表面。

图4 (a~c)NbFe0和(d~f)NbFe0.10样品的TEM和HRTEM图;(g、h)NbFe0.10样品的EDS谱图;(i~k)NbFe0.10样品的HRTEM-映射图Fig.4 TEM and HRTEM images of(a~c)NbFe0 and(d~f)NbFe0.10;(g,h)EDS spectrum of NbFe0.10;(i~k)HRTEM-mapping images of NbFe0.10
三维海胆状结构一般具有较大的比表面积。从图5a可以看出,所得样品的N2吸附-脱附等温线与Ⅳ型等温线相对应,滞后环属于H3型,说明合成的催化剂具有介孔结构,这些介孔主要来自于海胆状纳米球表面垂直生长的纳米棒之间的缝隙。图5b是样品的BJH孔径分布图,由图可知,所有样品的孔径分布较为集中:NbFe0样品的孔径为5.6 nm,当引入Fe3+后样品的孔径减小为4.8~5.0 nm,说明表面纳米棒在受到Fe3+影响不能长长的时候变得略微密集,导致纳米棒之间的缝隙变小,催化剂的孔容也相应地逐渐减小(图5b插图)。此外,样品的BET比 表 面 积 分 别 为 147 m2·g-1(NbFe0)、168 m2·g-1(NbFe0.05)、161 m2·g-1(NbFe0.10) 和 149 m2·g-1(NbFe0.20)。引入少量Fe3+(x=0.05)后,样品的比表面积显著增加(图5b插图),这是因为少量Fe物种的修饰使得样品颗粒尺寸减小,分散更加均匀。随着Fe3+引入量的增加(x=0.10),样品直径继续减小,发生团聚,比表面积较NbFe0.05有所减小。当x=0.20时,纳米球直径增加,使其比表面积继续减小,但是引入Fe3+后样品的比表面积均大于NbFe0样品的比表面积。由于多相光催化反应发生在光催化剂的表面,催化剂比表面积越大,对于染料分子的吸附能力越强,光催化效率越高。因此,Fe物种修饰的NbFex样品的大比表面积将有助于增强其光催化活性。

图5 NbFex样品的(a)N2吸附-脱附等温线和(b)BJH孔径分布及BET比表面积和孔容(插图)Fig.5 (a)N2adsorption-desorption isotherms and(b)BJH pore size distribution plot,BET specific surface areas and pore volumes(inset)of all NbFex samples
对NbFe0和NbFe0.10样品进行XPS分析,结果如图6所示。NbFe0.10的XPS全谱图(图6a)证明了合成产物中Fe、O、Nb元素的存在。对Fe2p结合能进行分峰处理(图6b)发现,NbFe0.10中有Fe2+和Fe3+两种价态出现,两者的面积比为1∶1,即物质的量之比为1∶1,然而在XRD图中只出现了α-Fe2O3的衍射峰。Ribeiro等[26]采用共沉淀法制备了Nb-Fe光催化剂,并对其结构进行了解析,发现经500℃空气焙烧后Fe物种包含α-Fe2O3、FeNb2O6和Fe4Nb2O9。因此,我们合成的样品经500℃空气焙烧后Fe2+物种极有可能是以上2种铌酸亚铁,在此记做Fe(Ⅱ)NbxOy。XRD图中无法检测到Fe(Ⅱ)NbxOy的存在可能是由于其颗粒尺寸较小且结晶度低[27]。图6c是O1s的分峰拟合谱图,O1s的谱峰来自晶格氧(530.18 eV)、表面羟基(531.03 eV)和表面吸附水(532.25 eV)三部分的贡献。在NbFe0.10样品中的表面羟基数相对NbFe0样品略有增加,主要原因是其结晶度稍有降低。图6d是Nb3d的分峰拟合谱图,可以看到,NbFe0和NbFe0.10样品的Nb3d峰位置和峰型几乎重合,其中Nb3d5/2的结合能约为207.11 eV,Nb3d3/2的结合能约为209.85 eV,表明Nb以+5价存在[28],不存在明显的氧缺陷,即Fe3+的引入没有对Nb2O5的配位环境造成太大影响。

图6 NbFe0.10的(a)XPS全谱图和(b)Fe2p谱图;NbFe0和NbFe0.10的(c)O1s和(d)Nb3d谱图Fig.6 (a)XPS survey spectrum and(b)Fe2p spectrum of NbFe0.10;(c)O1s and(d)Nb3d spectra for NbFe0 and NbFe0.10
2.2 光催化降解类吩噻嗪染料性能
图7是NbFex样品的UV-Vis吸收谱图和禁带宽度(Eg)图。Eg是衡量半导体性质的一个重要参数,通常可以用切线外延的Tauc法得到Eg值[19]:以[F(R∞)hν]m对hν作图,其中F(R∞)、h、ν、m分别是吸光系数、普朗克常量、光频率和半导体的固有性质,对于间接半导体 Nb2O5,m 为 1/2。F(R∞)可以根据 Kubelka-Munk函数从UV-Vis吸收曲线中得到:F(R∞)=(1-R∞)2/(2R∞),R∞是相对漫反射率,R∞与吸光度 A 有以下关系:
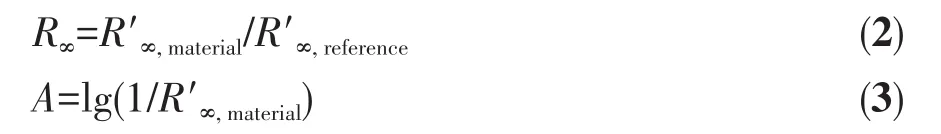
其中,R′∞,reference、R′∞,material分别是参比材料和测试样品的绝对漫反射率,实验中BaSO4是参比材料,其R′∞,reference接近于1,所以R∞=R′∞,material。从图7a中可以看出纯Nb2O5样品只在紫外光区有强吸收,而表面复合Fe物种(Fe2O3和Fe(Ⅱ)NbxOy)后,样品的光吸收发生红移,且随Fe含量的增加红移程度增加,这是因为Fe物种的引入改变了Nb2O5的尺寸、表面结构及组成。对Tauc曲线中最陡的位置作斜率可以得到 NbFex样品的 Eg值(图 7b),分别为 3.26 eV(Nb-Fe0)、3.06 eV(NbFe0.05)、2.86 eV(NbFe0.10)和 2.80 eV(NbFe0.20)。因此,在Nb2O5表面通过一步法原位复合Fe物种可以有效减小样品的Eg值。
图7c是NbFex样品在激发波长为315 nm下的PL谱图,所有样品在402、467和625 nm处均有发射峰,其中402和467 nm处的发射峰来自光生电子和空穴的复合[15],而625 nm处的峰则归属于带隙间的局域态[19]。从图中可以清楚地看到,没有Fe3+作用的NbFe0样品具有很高的光生电子和空穴复合率,而形成的Fe物种可以有效降低Nb2O5光生载流子的复合,且随着Fe含量增多效果更明显,这为进一步提高海胆状Nb2O5纳米球光催化活性提供了可能。
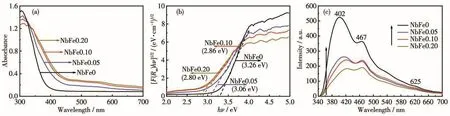
图7 (a)NbFex样品的UV-Vis吸收谱图;(b)Tauc曲线;(c)NbFex样品的PL谱图(激发波长为315 nm)Fig.7 (a)UV-Vis absorption spectra of NbFex;(b)Tauc plots;(c)PL spectra of all NbFex samples(excitation wavelength was 315 nm)
将4个不同nFe/nNb的样品用于全光谱降解类吩噻嗪染料MB。在混合0.1 mL H2O2(30%)的条件下,其降解过程的UV-Vis吸收谱图及浓度-时间曲线如图8所示。相对于NbFe0样品(图8a),引入Fe物种后样品展现出了较高的光催化活性(图8b~8d)。在纯H2O2作用下,染料降解效率很低,将催化剂加入体系之后降解效率迅速升高(图8e)。20 min后,NbFe0样品降解了57%的染料,但NbFe0.20已经完成了85%的降解,NbFe0.05与NbFe0.10样品的降解效率均达到90%左右,所以此过程中H2O2自身降解的贡献可以忽略不计。此外,表观速率常数k可以用来定量、直观地获得催化剂的光催化反应活性,计算公式:-ln(c/c0)=kt,结果如图8f和表1所示,NbFe0.10样品具有最大的k值(0.102 9 min-1),此值是NbFe0样品(0.051 3 min-1)的2倍。催化剂单位面积上的反应速率常数ks更能体现催化剂的催化活性,因此使用各NbFex催化剂的比表面积归一化速率常数得到ks(表1)。可以看出NbFe0、NbFe0.05、NbFe0.10、NbFe0.20的ks成递增趋势,说明 Fe物种的引入确实可以提高Nb2O5的光催化活性。此外,我们考察了纳米Fe2O3(合成方法参考Liu等[29]工作)的光催化降解MB的性能,如图9所示,其降解活性很低。因为单一Fe(Ⅱ)NbxOy(FeNb2O6或Fe4Nb2O9)很难得到,所以在文中没有纯Fe(Ⅱ)NbxOy对应的数据。没有H2O2参与时的NbFe0.10活性同样展示在图9中,可以看出此时NbFe0.10几乎没有光催化活性。因此,Fe物种和H2O2对于Nb2O5光催化降解性能的提高作用必不可少(Fe物种和H2O2的具体作用见后文分析)。
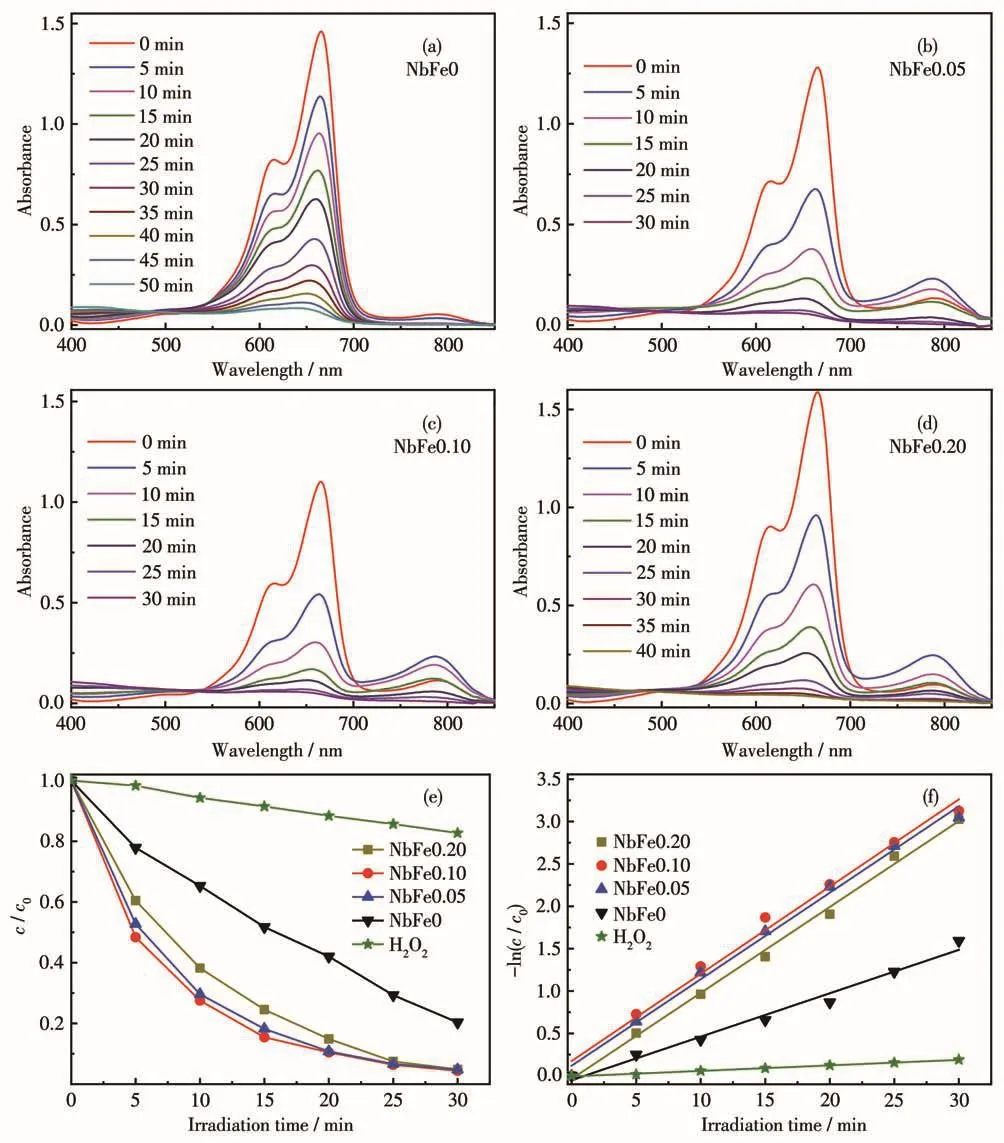
图8 NbFex样品(a~d)光催化降解MB(20 mg·L-1)的UV-Vis吸收谱图、(e)浓度-时间曲线及(f)动力学曲线Fig.8 (a~d)UV-Vis absorption spectra for degradation of MB(20 mg·L-1),(e)corresponding concentration-time curves and(f)kinetic curves by using all NbFex samples

图9 不同催化剂的(a)浓度-时间降解曲线及(b)对应的表观速率常数k(MB:20 mg·L-1)Fig.9 (a)Concentration-time curves and(b)apparent rate constant k(MB:20 mg·L-1)over different catalysts
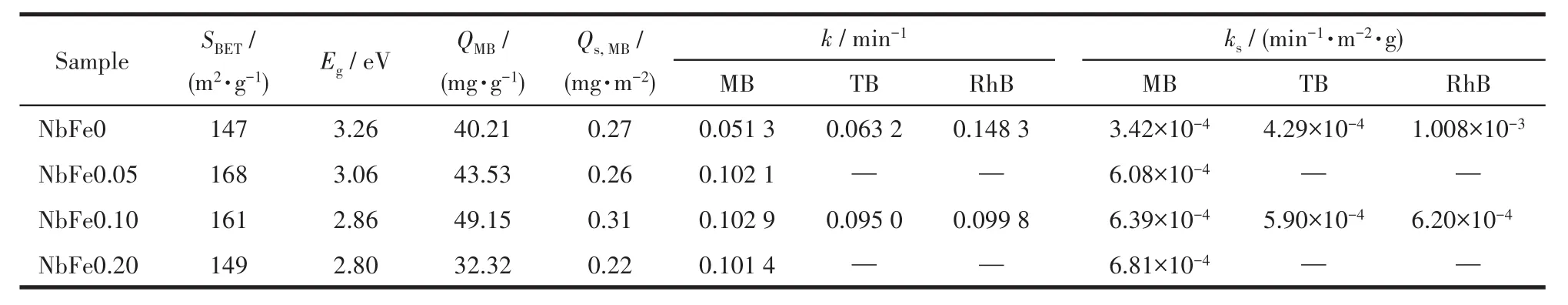
表1 NbFex的比表面积(SBET)、禁带宽度(Eg)、吸附量(QMB、Qs,MB)和速率常数(k,ks)Table 1 Specific surface area(SBET),band gap(Eg),adsorption capacity(QMB,Qs,MB)and reaction rate constant(k,ks)of NbFex
众所周知,光降解的第一步是催化剂对染料的吸附,即使是高活性催化剂,也只有足够的染料吸附到催化剂表面才能发挥其优势。因此,我们计算了NbFex对MB的吸附量(QMB),结果如表1所示,将其归一化得到单位面积吸附量Qs,MB后发现,NbFe0.10对MB的单位面积吸附量最大,说明Fe含量适中的情况下可以促进Nb2O5催化剂对MB的吸附能力。此外,我们选择了另外一种和MB具有相似结构的类吩噻嗪染料TB和具有不同结构的染料RhB(图S1),用NbFe0和NbFe0.10进行了其降解活性实验,结果如图10及图S2所示。可以发现,NbFe0和NbFe0.10样品光催化降解TB的结果与降解MB的实验相似,NbFe0.10的活性(k=0.095 0 min-1,ks=5.90×10-4min-1·m-2·g)大于 NbFe0 的活性(k=0.063 2 min-1,ks=4.29×10-4min-1·m-2·g)(表 1)。然而在RhB的光催化降解实验中,含Fe样品NbFe0.10的光催化活性(k=0.099 8 min-1,ks=6.20×10-4min-1·m-2·g)却比纯 Nb2O5样品活性(k=0.148 3 min-1,ks=1.008×10-3min-1·m-2·g)低。结合 NbFe0.10 降解 MB 的结果,表明Fe物种修饰的Nb2O5催化剂对类吩噻嗪染料具有明显的选择性降解特性。计算NbFe0和NbFe0.10对TB和RhB的吸附量QTB、QRhB及相应的Qs,TB、Qs,RhB,结果如图11所示。可以看出在引入Fe3+之后,催化剂对MB和TB的单位面积吸附量增加,然而对RhB的吸附量反而减少。主要原因是Fe物种可以和类吩噻嗪染料分子中N、S键形成配位吸附,从而增加了催化剂对染料的吸附能力,进而保证了后续的光催化降解过程,这一现象在我们的前期工作中已经有了详细说明[2,19]。
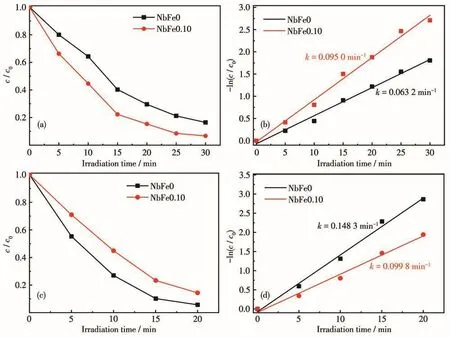
图10 NbFe0和NbFe0.10光催化降解(a、b)TB和(c、d)RhB性能(20 mg·L-1)Fig.10 Photocatalytic degradation of(a,b)TB and(c,d)RhB by NbFe0 and NbFe0.10(20 mg·L-1)
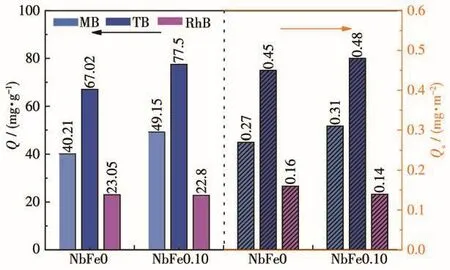
图11 NbFe0和NbFe0.10对MB、TB和RhB的吸附量Fig.11 Adsorption amount of MB,TB and RhB on NbFe0 and NbFe0.10
在光降解阶段,海胆状Nb2O5纳米球表面原位复合的Fe物种可以充当Nb2O5导带光生电子的俘获井,进而通过Fe3++e-→Fe2+的还原消耗掉光电子,降低Nb2O5光生电子和空穴的复合[18-20]。Nb2O5导带电子分离使其价带空穴更容易氧化吸附在表面的有机污染物。同时,Fe2+和H2O2之间发生Fenton反应,产生大量·OH用于染料的氧化,此过程中Fe2+再一次被氧化为Fe3+,反应继续循环[19]。此外,将参与降解的NbFe0.10催化剂离心分离,300℃焙烧2 h,在相同降解条件下继续使用,3次循环后催化剂仍具有良好的光催化降解活性,结果如图S3所示。
2.3 光催化降解机理
利用经验方程,可以从理论上合理地预测半导体的导带(CB)和价带(VB)位置,公式如下:

其中,X为半导体的绝对电负性,由其组成原子的电负性的几何平均值得到(Nb2O5的X值为6.29);Eg是半导体的禁带宽度;Ee是氢原子上自由电子的能量,一般取值为4.5 eV。实验所得纯Nb2O5的Eg=3.26 eV,所以计算得到 Nb2O5的 ECB=0.16 eV,EVB=3.42 eV。此外,根据文献报道可知,α-Fe2O3纳米团簇或者纳米小颗粒的CB位置在0.46~0.77 eV之间,Eg约为2.2 eV[18-19];虽然我们不能明确得到Fe(Ⅱ)NbxOy的Eg和ECB的具体数值,但是从Ribeiro等[26]的工作中可以看出,含有Fe(Ⅱ)NbxOy组分的催化剂的UV-Vis吸收谱图具有明显的可见光吸收特点,因此不难猜测Fe(Ⅱ)NbxOy的 Eg不会大于纯 Nb2O5的 Eg值。此外,铌酸亚铁的CB位置一般会降低[19],且根据Bi和Xu[20]的研究可知,即使Fe(Ⅱ)NbxOy的CB位置高于Nb2O5的CB位置,在光降解过程中,基底Nb2O5上激发的电子也会向Fe(Ⅱ)NbxOy能带较低的位置迁移,从而促进其光生电子的分离。因此,Fe物种修饰的Nb2O5光催化降解过程如下:催化剂NbFex(x=0.05、0.10、0.20)在光照和H2O2的作用下将有机污染物降解为CO2和H2O,而其本身不会发生变化(总反应式6)。催化剂基底Nb2O5受光激发产生电子和空穴(式7),电子传递到 Fe2O3和Fe(Ⅱ)NbxOy的 CB上;迁移到Fe物种CB上的光生电子促进还原反应的发生(式8);随后Fe2+和H2O2之间发生Fenton反应,产生大量·OH,此过程中Fe2+再一次被氧化为Fe3+,使反应开始循环(式9);同时,Nb2O5的VB上的空穴氧化H2O产生·OH和H+(式10);Fenton反应和空穴产生的·OH同时氧化染料产生CO2和H2O(式11),整个过程示意图如图12所示。需要注意的是,Fe2O3和Fe(Ⅱ)NbxOy在样品中的含量很少,且自身产生的电子和空穴易复合,所以其本身对染料的光催化降解反应贡献很小。
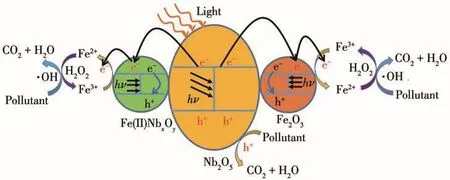
图12 Fe物种修饰的Nb2O5光催化降解有机染料机理Fig.12 Proposed mechanism of photocatalytic degradation of organic dyes by Fe species modified Nb2O5catalyst under full light irradiation
总反应:
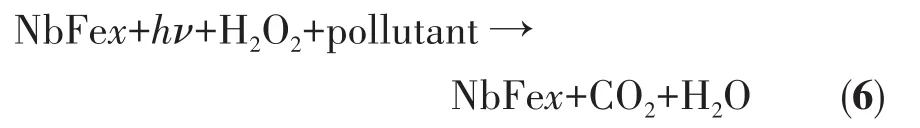
分步反应:


3 结 论
在水热法合成海胆状Nb2O5纳米球的基础上,以硝酸铁为铁源,通过向体系直接引入Fe3+成功制备了Fe物种修饰的海胆状Nb2O5纳米球。实验发现,引入Fe3+没有显著改变Nb2O5的微观形貌和晶体结构,生成的Fe物种以低结晶度的Fe2O3和Fe(Ⅱ)NbxOy分布在Nb2O5纳米球表面。在光催化过程中,Fe物种通过与Nb2O5导带匹配和快速发生Fenton反应可以有效分离Nb2O5导带上的光生电子,降低载流子的复合,同时产生大量·OH用于染料氧化。此外,原位复合的Fe物种可以有效吸附含N和S的类吩噻嗪染料,所以催化剂对此类染料表现出了明显的选择性降解作用,且催化剂易于回收再利用。
Supporting information is available at http://www.wjhxxb.cn
