1-苯基-3-甲基-4-苯甲酰基-5-吡唑啉酮镝(Ⅲ)配合物的晶体结构、磁性及理论分析
2021-04-10景佳慧孟银杉张义权陆海全刘姗姗
景佳慧 刘 斌 孟银杉 张义权 陆海全 刘姗姗*,
(1北京石油化工学院化学工程学院,燃料清洁化及高效催化减排技术北京市重点实验室,北京 102617)
(2大连理工大学,精细化工国家重点实验室,大连 116024)
(3北京大学化学与分子工程学院,北京分子科学国家实验室,稀土材料化学及应用国家重点实验室,北京 100871)
(4南京师范大学,江苏省大规模复杂系统数值模拟重点实验室,南京 210023)
0 引 言
信息技术的快速发展对存储材料的体积、存储密度等提出了更高的要求。单分子磁体可以单个分子为磁记忆单元,有望应用于高密度磁存储器等领域。近年来单分子磁体的研究已经取得了重要突破,磁阻塞温度可达到80 K[1]。提高单分子磁体的磁阻塞温度为该领域不断追求的目标,而多项实验研究和理论分析指出了设计性能优异单分子磁体的策略[2-12],如高对称性、直线型二配位的镝配合物[1,4,13-15]。同时,研究表明镝配合物的磁各向异性与配位环境中负电荷密度的分布情况密切相关[16]。配位空间中负电荷密度分布呈轴向性时,该镝配合物将会表现出更强的磁各向异性,而配体所连基团的电子效应会影响负电荷密度的分布。由此可见,电子效应对磁各向异性影响的研究具有重要意义。
在已报道的单分子磁体类型中,氧基配体构筑的稀土配合物种类丰富,如β-二酮类配体、醇类配体、烷氧基类配体等[17-19],其中β-二酮类配体应用广泛,如乙酰丙酮、二苯甲酰甲烷、丁二酮、庚二酮及其衍生物等[20-23]。此外,吡唑啉酮类β-二酮配体已被广泛应用在金属离子分离领域、发光领域和生物活性领域[24-26]。我们以1-苯基-3-甲基-4-苯甲酰基-5-吡唑啉酮(Hpmbp)配体构筑了一例单核镝配合物[Dy(pmbp)3(H2O)2]·CH3CN(1),表征了该配合物的单分子磁体性能,通过理论计算分析了其磁易轴方向并研究了pmbp-配体中共轭效应对磁各向异性的影响。
1 实验部分
1.1 主要试剂与仪器
本实验所涉及操作在空气氛围中完成,所用试剂均未经纯化直接使用。元素分析测试在Elementar Vario EL CUBE(德国)元素分析仪完成。粉末X射线衍射数据在Rigaku(日本理学)DMAX粉末X射线衍射仪上使用Cu Kα(λ=0.154 06 nm)在40 kV和40 mA下获得,扫描范围为5°~50°。样品的直流磁性数据和交流磁性数据测试分别在Quantum Design MPMS-XL-7磁测量仪和PPMS-9上进行。
利用MOLCAS 8.4程序包[27]和SINGLE_ANISO程序[28-30]进行CASSCF计算得到了能级、g张量、mJ值和磁轴等信息。
1.2 配合物[Dy(pmbp)3(H2O)2]·CH3CN(1)的合成
称取Hpmbp 0.843 2 g和氢氧化钠0.152 8 g于50 mL蒸馏水中,加热搅拌溶解,趁热过滤,将滤液缓慢滴入0.439 2 g Dy(NO3)3·5H2O的100 mL蒸馏水溶液中,产生浅黄色的粉末。再加入100 mL蒸馏水,抽滤,水洗3次,无水乙醇洗1次,将粉末在80℃下干燥1.5 h,得粉末0.812 9 g。称取0.199 2 g粉末溶于5 mL乙腈和5 mL丙酮溶液中进行重结晶,过滤,静置培养单晶,几天后得到黄色晶体,产率43.2%。元素分析按C53H46DyN7O8计算的理论值(%):C 59.41,H 4.33,N 9.15;实验值(%):C 59.17,H 4.28,N 9.10。
1.3 配合物的晶体结构测定
选取大小为0.41 mm×0.05 mm×0.04 mm的单晶置于安捷伦SuperNova Dual Atlas CCD单晶衍射仪上,采用 Mo Kα(λ=0.071 073 nm)射线作为光源,在180(2)K下收集衍射数据。结构使用SHELXL解析[31],水上的氢原子由差值傅里叶合成图得到,其他氢原子由理论加氢得到。配合物1的晶体学数据列于表1,主要键长和键角列于表2。

表1 配合物1的晶体学数据Table 1 Crystal data and structure refinement for complex 1

表2 配合物1的部分键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and angles(°)for complex 1
CCDC:2032517。
2 结果与讨论
2.1 配合物1的晶体结构
X射线单晶衍射数据(表1)表明配合物1结晶于单斜晶系P21/n空间群。如图1所示,Dy(Ⅲ)离子与3个pmbp-配体和2个H2O分子配位。此外,还有一分子乙腈未参与配位。Dy(Ⅲ)离子为[DyO8]型八配位构型,其中6个O原子来自3个pmbp-配体,2个O原子来自2个H2O分子。Dy—O(pmbp-)平均键长(0.235 1 nm)明显短于Dy—O(H2O)平均键长(0.241 9 nm)。在配合物结构中,2个H2O分子相邻,2个pmbp-配体处于对位,另一个pmbp-配体与2个H2O分子处于对位。O7—Dy1—O8键角为71.90(11)°,pmbp-中2个O原子与Dy(Ⅲ)的键角分别为73.81(13)°、73.78(11)°和73.46(12)°。以其中一个配位的pmbp-配体为例,分析pmbp-内的电子共轭效应。C34所在的苯环与螯合六元环和吡唑环的二面角为52.478°和65.778°,接近于垂直关系,且C34—C28键长为0.150 7 nm,与C—C单键键长相符,表明苯环与螯合六元环和吡唑环未形成共轭;吡唑环与C24所在苯环的二面角为24.001°,具有一定程度的共轭,该部分结构特点与[La(pmbp)3(H2O)2]、[Sm(pmbp)3(H2O)2]相似[32-33]。用SHAPE程序[34]计算得出配合物1中Dy(Ⅲ)离子的配位环境接近于四方反棱柱构型(偏离度0.731)。相邻Dy(Ⅲ)离子之间的最短距离为0.996 8 nm。

图1 配合物1的结构及O1~O8的Mulliken电荷(a)和Dy(Ⅲ)离子的配位环境(b)Fig.1 Structure of complex 1 and Mulliken charges of O1~O8(a)and coordination environment of Dy(Ⅲ) ion(b)
2.2 配合物1的粉末衍射测试
在室温下对配合物1进行了粉末X射线衍射测试。如图2所示,实验数据与由单晶结构数据模拟所得数据相符,表明样品纯度符合后续测试要求。
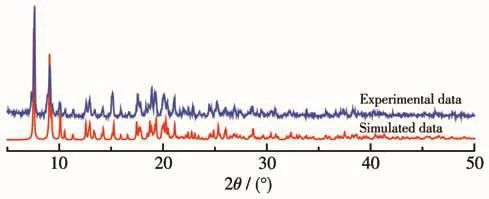
图2 配合物1的粉末X射线衍射图Fig.2 Powder X-ray diffraction pattern of complex 1
2.3 配合物1的磁性
我们测量了配合物1在4~300 K的温度范围内及1 kOe外加磁场中的变温磁化率。如图3所示,在300 K时配合物1的χMT值为13.90 emu·K·mol-1,接近于一个自由Dy(Ⅲ)离子的理论值14.17 emu·K·mol-1。随着温度的降低,χMT值平缓下降;当温度达到4 K时,χMT值降到最小值:10.98 emu·K·mol-1,这可归因于Stark能级的退占据。在0~50 kOe的磁场范围内,在2 K下测量了配合物1的饱和磁化强度曲线(图 4a),其最大值为 4.99Nβ,低于理论值 10Nβ。同时,不同温度(2、3、5、8、10 K)下的磁化强度曲线(图4b)不能重叠,表明配合物1存在磁各向异性。
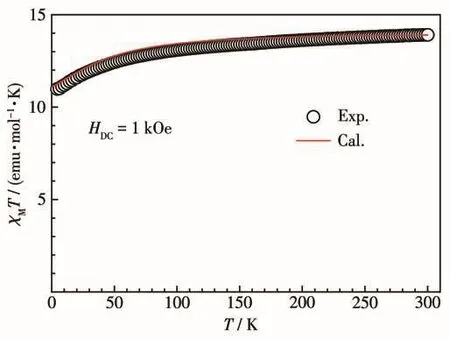
图3 配合物1的变温磁化率曲线:实验测量(黑色)和从头计算(红色)Fig.3 Temperature-dependent magnetic susceptibility of complex 1 by experimental measurements(black)and ab initio calculations(red)
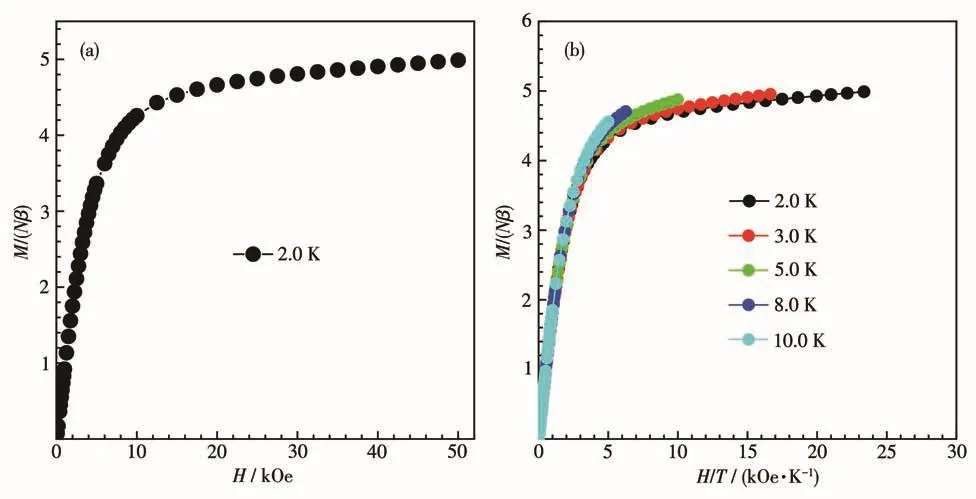
图4 配合物1在2 K下的磁化强度随磁场变化曲线(a)和M-H/T曲线(b)Fig.4 Magnetic field dependence of magnetization at 2 K(a)and M-H/T plots(b)for complex 1
为研究配合物1的磁弛豫行为,对其进行了2~10 K温度区间的交流磁化率随频率(100~10 000 Hz)的测量。在零场下可以观察到虚部交流磁化率信号,但是并没观察到χ″的峰值,表明存在强的量子隧穿(QTM)效应(图5a)。在2 kOe外加磁场下,QTM效应被抑制,可以观察到χ″峰值(图5b)。随着温度的升高,χ″峰逐渐向高频方向移动,表明存在慢的磁弛豫行为。以 Arrhenius公式 τ=τ0exp[ΔE/(kBT)]对ln τ-T-1图(图6)进行有效能垒拟合,得出了配合物1在2 kOe直流场下的能垒为42 K,指前因子τ0=2.07×10-7s。

图5 配合物1在零直流场下的虚部交流磁化率曲线(a)和在外加2 kOe直流场下的虚部交流磁化率曲线(b)Fig.5 Frequency dependence of χ″ac susceptibilities in the absence of dc field(a)and under a 2 kOe dc field(b)for complex 1
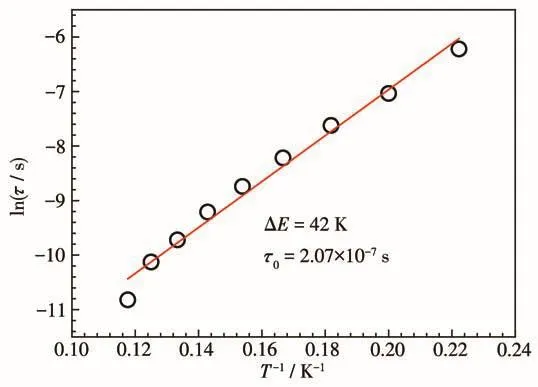
图6 配合物1的有效能垒拟合图Fig.6 Effective energy barrier plot of complex 1
2.4 理论分析
为了进一步研究配合物1的磁各向异性和微观电子能级态的组成,我们利用MOLCAS 8.4软件的CASSCF/RASSI-SO/SINGLE_ANISO程序[27]对配合物1进行从头计算。磁性测量和理论计算得到的变温磁化率曲线,可以很好地吻合(图3)。表3为配合物1的微观电子能级态 8个KDs和对应的 g(gx、gy、gz)。基态gz值为18.818,严重偏离Dy(Ⅲ)离子的Ising极限值20;gx和gy值的大小与横向磁各向异性相关,理论研究认为gx、gy>0.015的情况下,配合物不容易在零场下表现出慢的磁弛豫行为[16],而配合物1的基态微观电子能级的gx和gy值远大于该值。同时,微观电子能级基态横向磁矩(0.067μB)较大,表明存在强的QTM过程。以上均导致配合物1在零场下难以表现出明显的慢磁弛豫行为。此外,基态波函数中|±15/2〉仅占84.33%,也说明了这一问题。
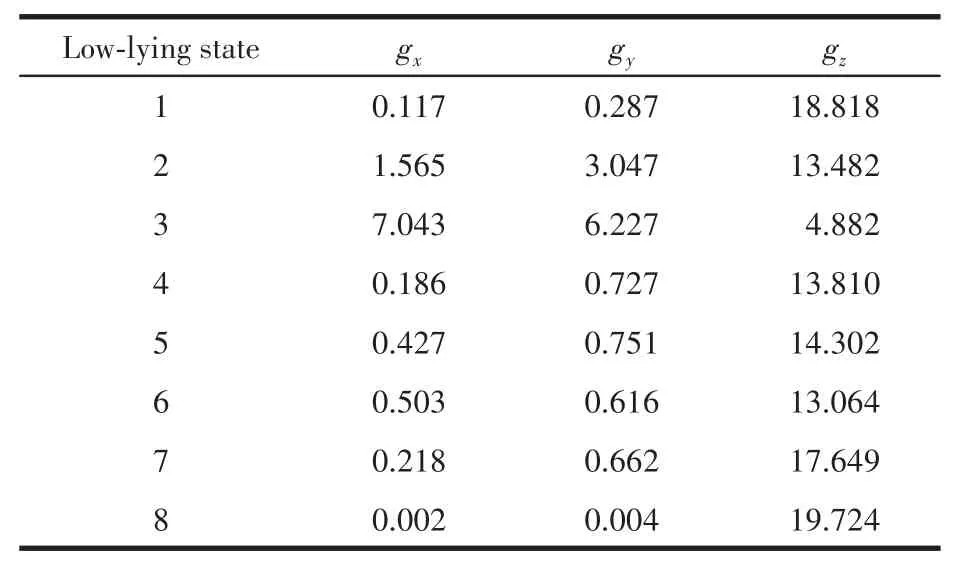
表3 配合物1的八个最低态的g值Table 3 g values for eight low-lying states of complex 1
理论计算所得微观电子能级基态磁各向异性轴沿着2个pmbp-配体方向(图7),与Dy—O3和Dy—O4 的夹角为 35.442°和 38.604°,近似处于 pmbp-配体2个O原子中间位置。在配合物1的结构中,其中2个pmbp-配体处于对位,另一个pmbp-配体与2分子水处于对位。理论计算得出O1~O8的Mulliken电荷如图1a所示,由此可见沿着2个对位pmbp-配体方向的负电荷密度更集中,相应的静电势更强,因而磁易轴沿着该方向更稳定。

图7 配合物1的磁易轴方向Fig.7 Orientation of magnetic easy axis of complex 1
由磁性表征结果及理论计算可知该配合物磁各向异性并不强,这与其结构密切相关。分析其结构可知,配体pmbp-中螯合六元环与吡唑环及相连苯环存在一定程度的共轭效应,电子的离域导致配位氧原子上的负电荷分散至吡唑环及相连苯环。磁易轴方向2个pmbp-配体的负电荷离域不利于|±15/2〉态的稳定,因此难以构筑强的磁各向异性。
3 结 论
我们以β-二酮配体1-苯基-3-甲基-4-苯甲酰基-5-吡唑啉酮合成了一例单核稀土配合物[Dy(pmbp)3(H2O)2]·CH3CN,并对其进行了元素分析、单晶X射线衍射及磁性表征。Dy(Ⅲ)离子处于畸变的四方反棱柱配位环境中。该配合物在2 kOe的外加磁场下可以表现出慢的磁弛豫行为,为一例场诱导单分子磁体,其能垒为42 K,τ0=2.07×10-7s。理论计算得出其磁易轴方向沿着对位的2个pmbp-配体。同时,微观电子能级g值及基态横向磁矩大小解释了该配合物在零场下难以表现出慢的磁弛豫行为的现象,磁构关系分析表明这与pmbp-配体中的共轭效应相关。
