阻塞性睡眠呼吸暂停低通气综合征导致心血管疾病相关机制
2021-04-09郭子宏
冯 葶 郭子宏
阻塞性睡眠呼吸暂停低通气综合征[1,2](Obstructive sleep apnea hypopnea syndrome,OSAHS)是睡眠中出现上气道完全或部分阻塞,引起呼吸暂停及低通气反复发作30次以上,或呼吸暂停低通气指数(apnea-hypopnea index,AHI)≥5次/h,产生慢性间歇性缺氧、二氧化碳潴留等,导致睡眠节律紊乱、白天嗜睡,引起或加重高血压、冠心病、糖尿病、心律失常、心力衰竭、脑卒中等,甚至猝死的一种全身性疾病,常伴发多系统损害。
据流行病学统计,中年男性阻塞性睡眠呼吸暂停(Obstructive sleep apnea,OSA)患病率为22%(9%~37%),女性为17%(4%~50%),且随年龄增加和肥胖程度加重而增加[3]。Senaratna CV[4]等的人群患病率研究结果显示,在AHI≥5次/h时,总体人群患病率约9%~38%,在AHI≥15次/h时,普通成人人群患病率约6%~17%,高龄人群高达49%。
近年来,多项研究证明[5~7],OSAHS与心血管疾病具有显著相关性,独立于肥胖、年龄、吸烟等危险因素,且发病率与AHI成正相关[8]。本文综合近年来国内外研究进展,从OSAHS诱发或加重心血管疾病的病理生理机制方面进行综述。
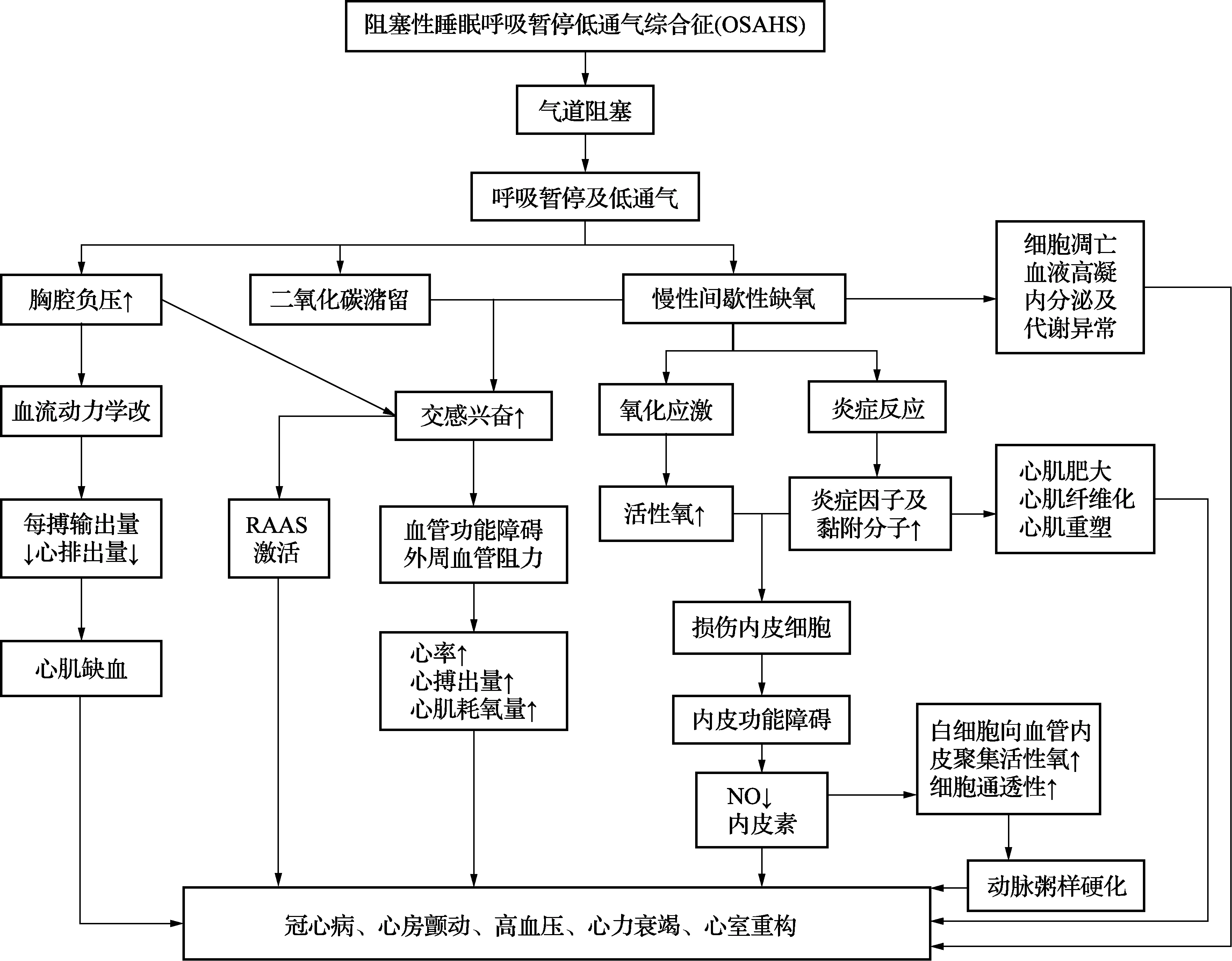
图1 OSAHS诱发或加重心血管疾病的相关病理生理机制
1. OSAHS诱发或加重心血管疾病的相关机制
OSAHS诱发或加重心血管疾病的机制十分复杂,目前尚不完全明确。潜在机制可能是气道阻塞引起呼吸暂停及低通气,产生慢性间歇性缺氧,导致自主神经功能紊乱[7]等,从而继发机体病理生理改变。这些机制途径对于冠心病、高血压、心律失常、心衰等的发生发展至关重要[5,9]。OSAHS诱发或加重心血管疾病的相关病理生理机制如图1所示。
1.1 胸腔负压变化 OSAHS患者睡眠时上气道完全或部分阻塞,引起呼吸暂停及低通气反复发作,机体为对抗气道阻塞,胸腔内负压迅速升高可达-80cmH2O[10],并随呼吸程度的增加而加剧。这一压力改变一方面导致心脏内外压力差及左心室透壁压增大,刺激交感神经兴奋[11],同时增加了左心室及主动脉的跨壁压力[10],降低了心室功能、血流动力学及自主神经功能稳定性;另一方面[12~14],胸腔负压迅速增大可使左房容量减少,左室收缩末期容积急剧增加,左室后负荷增加,导致左心室射血分数、心排血量减少,从而减少了冠脉血流,导致心肌缺血。此外,胸腔压力反复变化、回心血量波动刺激主动脉体和颈动脉窦压力感受器,刺激交感神经兴奋;长期反复心肌缺血、交感神经兴奋可引发心衰,而缺氧诱发的肺血管收缩可导致肺动脉高压,加重右室后负荷,促进心衰的发展[12,13]。
1.2 自主神经功能紊乱 OSAHS呼吸暂停反复发生引起的间歇性缺氧和高碳酸血症可作用于交感神经调节中枢,并增加外周化学感受器的敏感性,从而激活交感神经系统[15],并抑制迷走神经张力,每晚重复上述刺激过程,最后使交感神经兴奋状态在日间持续存在[1]。这种自主神经功能紊乱使得血管收缩和舒张功能障碍,外周血管阻力增加,引起血压升高、心率增快、心排血量减少、心肌耗氧量增加、冠状动脉供血量减少,从而诱发心肌缺血,促进冠心病的发生;还可引起难治性高血压、心肌重塑、心衰、肺动脉高压等疾病的发生。此外,研究发现急慢性呼吸暂停发作对房颤的发作、维持及复发均起着促进作用[16],OSA动物模型[17]及急性间歇性缺氧动物模型研究[18]均得出相同结论,提示对心脏自主神经系统的调节可能是治疗OSAHS诱导房颤的一种新疗法。
1.3 氧化应激、炎症反应及内皮损伤 OSAHS呼吸暂停及低通气反复发作引起二氧化碳潴留、高碳酸血症,这种重复缺氧-再氧合的慢性间歇性缺氧(chronic intermittent hypoxia,CIH)模式类似缺血-再灌注损伤,可诱导机体氧化应激及炎症系统激活,产生大量活性氧(ROS),同时CIH可激活核因子-κB等介导的炎性途径,通过血管黏附分子-1(VCAM-1)、细胞间黏附分子-1(ICAM-1)等黏附分子介导,使白细胞黏附,促进或加重血管内皮损伤[19,20]。ROS[21]一方面可通过启动炎症联级反应致促炎症因子(如TNF-α、IL-6、CRP等)及黏附分子过度表达,产生大量炎症细胞因子,引起炎症反应;另一方面促进炎症细胞的生长和迁移、激活转录因子,损伤内皮细胞和血管壁,引起内皮功能障碍。CIH还可以引起NO水平下降,内皮素增加,促进血管收缩,血压升高,NO下降又可引起血小板聚集、白细胞黏附,促进ROS生成。而内皮细胞受损又可刺激炎症因子释放,诱导炎细胞向血管内皮聚集,触发动脉粥样硬化发生,增加冠脉钙化[22]和急性冠状动脉综合征[23,24]的发生率,加速了心血管疾病的发展。此外,ROS过度产生可引发膜过氧化、酶及蛋白质变性和DNA突变,导致细胞功能异常,最终导致不可逆转的细胞损伤或死亡[25]。
研究发现,炎症因子在心肌重构的发展中起致病作用。IL-6和CRP可诱导心肌纤维化[26],TNF-α参与心肌细胞肥大、心脏重塑的调节,进而导致心室功能障碍[27]。有研究[28]证明CIH可引起大鼠心肌纤维化和心肌细胞肥大,并上调左心室心肌中IL-6、TNF-α等基因表达水平。这进一步说明OSAHS导致的CIH可引起心肌纤维化及心肌重构,从而诱发或加重高血压、房颤、心衰等心血管疾病。
大量研究表明[29~31]在CIH条件下,机体会产生各种炎症因子和细胞因子,激活氧化应激及炎症反应,引起内皮损伤。Tie YX[32]等证实CRP水平与OSAHS严重程度成正相关。Del Rio等人[33]发现,CIH增加颈动脉体中TNF-α和IL-1β水平,他们的另一项研究[34]发现颈动脉消融可减轻大鼠CIH引起的高血压和自主神经改变,表明CIH后高血压的维持依赖于CIH对颈动脉体的刺激,该研究为改善OSAHS相关性高血压的新疗法提供了实验依据。在一项针对OSAHS患者血浆中ICAM-1和VCAM-1水平的前瞻性研究[35]中,多因素logistic回归分析显示OSAHS与ICAM-1和VCAM-1高度表达独立相关,且与AHI成正相关。Fan Zi wen[36]等人发现内皮功能障碍与OSAHS的严重程度密切相关,且血清降钙素水平与内皮功能障碍密切相关,这表明在OSAHS患者出现临床症状之前,降钙素水平可作为评估内皮功能障碍发展的早期生物标志物,这为OSAHS患者内皮功能损伤的早期发现及随访观察提供了一种新型可靠的标志物。
1.4 细胞凋亡 截至目前,对于OSAHS诱导内皮细胞凋亡的具体分子机制尚不完全明确,有待进一步研究阐明。Kai-XiongLiu[37]等用CIH的细胞模型证明,CIH可以诱导microRNA的差异表达,且microRNA表达模式的改变与凋亡或自噬相关基因表达有关。为探讨OSAHS患者血管内皮细胞的损伤及凋亡机制,叶春幸[38]等人将OSAHS患者分为轻、中、重度及正常对照组,分离血清单核细胞(MNC),并与人脐静脉内皮细胞(HUVEC)共同培养48小时后,检测上清液中的TNF-α、IL-6及内皮细胞的凋亡率,结果发现轻、中、重度组MNC细胞产生TNF-α及IL-6均高于正常对照组(均P<0.01),且重度组MNC产生TNF-α及IL-6均高于轻、中度组(均P<0.01);轻、中、重度组内皮细胞的凋亡率较正常对照组显著升高(均P<0.01)。内皮细胞凋亡率与OSAHS患者AHI成正相关(r=0.58913,P=0.0106),与夜间最低血氧饱和度成负相关(r=-0.50737,P<0.01)。这说明OSAHS患者血管内皮损伤过程中,伴有内皮细胞凋亡,单核细胞在该过程中可能起促进作用,且这种作用与OSAHS严重程度、夜间缺氧密切相关。闫雅茹[39]等人发现,间歇低氧可通过TXNIP2介导线粒体功能障碍,导致线粒体依赖的凋亡蛋白释放,引起HUVEC的凋亡;而在间歇低氧处理24小时后再予线粒体保护剂Mito-TEMPO,线粒体相关凋亡蛋白释放减少;进一步证实了间歇性缺氧可引起血管内皮损伤,并诱导内皮细胞凋亡,而此过程可被Mito-TEMPO抑制,这为OSAHS相关性心血管疾病的精准治疗及靶向治疗提供了分子研究基础。
1.5 肾素-血管紧张素-醛固酮系统(RAAS)激活 OSAHS引起的交感神经激活、氧化应激、血管内皮损伤等均可刺激儿茶酚胺的释放,刺激机体肾素-血管紧张素-醛固酮系统(renin-angiotensin-aldosterone system,RAAS)激活,从而促进动脉粥样硬化、冠心病、高血压、心衰等的发生[19]。有证据表明[40],OSAHS患者中存在RAAS系统激活,且与AHI严重程度成正相关[41]。研究发现[42],高血压并发OSAHS的患者血浆醛固酮(Aldo)浓度明显升高,而血浆肾素活性(PRA)明显下降。一项荟萃分析[43]显示,OSA与较高的血管紧张素Ⅱ(AngⅡ)和Aldo水平相关,尤其在高血压患者中,且OSA至少部分通过刺激RAAS活性引起高血压。一项研究发现[44],Aldo水平较高的难治性高血压患者Aldo的升高可加重OSAHS的严重程度。但D.LYKOURAS[45]等人的研究得出相反的结论,发现中重度OSAH与非OSAH两组间的血清Aldo、肾素水平差异均无统计学意义(P=0.223,P=0.360),并得出结论,OSAH引起的CIH并未影响Aldo及肾素水平,OSAH与高血压的发展之间可能没有直接联系,而是通过交感神经系统的激活来建立的,闫晓娟[46]等人得出相同的结论。综上所述,RAAS激活可能是OSAHS诱导或加重心血管疾病的病理生理机制之一,但仍需多中心大样本研究来进一步证实。
1.6 血液的高凝状态 CIH刺激肾小球旁细胞生成促红细胞生成素,促进红细胞生成;此外,交感神经兴奋、RAAS激活、打鼾、张口呼吸使呼吸道水分蒸发增多,血容量减少,血液黏滞度增加;同时CIH刺激血浆纤维蛋白原水平升高、凝血因子活性增强[1,47];研究发现[48],中重度OSAHS凝血酶原时间(PT)和国际标准化比率(INR)与AHI成负相关,使得OSAHS患者处于血栓前状态,即血液呈不同程度的高凝、高聚状态,表现为血小板聚集和黏附功能增强、血浆纤维蛋白原浓度升高、纤维溶解系统活性减弱,这些因素相互作用,促进血栓的形成和AS的发展。
1.7 内分泌及代谢异常 OSAHS与代谢性疾病密切相关,患者血浆中甘油三酯、葡萄糖、胰岛素水平增高,胰岛素受体敏感性降低,导致胰岛素抵抗(IR),引起交感神经亢进、诱发炎症反应及内皮损伤,促进糖尿病、高血压、冠心病、肥胖等的发生[1,49]。研究发现[50],OSAHS患者发生糖代谢异常的风险是普通人的2.44倍,在排除肥胖干扰后,OSAHS血糖水平与AHI相关。然而,肥胖与OSAHS关系密切,体重降低10%会导致AHI发生26%~32%的变化[51],肥胖已成为OSAHS与心血管疾病之间的重要混杂因素。一项研究[52]发现CPAP治疗OSA可在短期内改善胰岛素敏感性,且该作用在中度肥胖者中维持了12周以上,提示OSA与IR之间可能存在因果关系;而S.R.Coughlin[53]等人随机盲法交叉试验结果不同,经过6周CPAP治疗后,OSA肥胖患者IR和血脂变化未见改善,这可能与治疗时间短和体重基线高有关[S.R.Coughlin等人的研究BMI基线为(36.1±7.6)kg/m2],提示可能存在肥胖的阈值水平,无论OSA存在与否或其严重程度如何,过量的体脂都是胰岛素敏感性的主要决定因素。另一项随机对照试验[54]发现,在对OSA分组进行减肥治疗和减肥联合CPAP治疗中,CRP、IR和血清甘油三酯水平降低,但仅接受CPAP治疗组未发现上述变化,与仅接受减肥治疗相比,联合治疗并无增量作用。这与另一项研究结果一致,在该研究中[55],即使在非肥胖患者中OSA也与炎症和IR显著相关,且CPAP不能改善炎症和IR。尽管目前还没有足够的临床证据支持OSAHS在IR和(或)血脂异常发展中的因果关系,但减轻体重对于降低该人群心血管疾病危险仍具有重要意义。
2.问题与展望
OSAHS是一种严重威胁人类健康的疾病,病因复杂,临床表现、并发症常累及多个系统,严重影响患者的生活质量,对其发生发展的认识和研究对于临床预防、诊断、治疗具有指导意义。提高临床医生对OSAHS的认识,对减少其对心血管系统及其他系统的损害具有重要的临床价值。同时,区别于传统的CPAP治疗,抑制RhoA/ROCK途径[28]、经导管去肾脏交感神经治疗[56]、颈动脉射频消融[34]、使用线粒体保护剂[39]等可作为OSAHS相关耐药性或难治性高血压、不能耐受CPAP治疗或治疗效果不理想的OSAHS相关性心血管疾病患者新的治疗选择。
