甘草苷体内暴露特征及体外跨膜转运机制研究
2021-04-07张爱杰刘羽康张英华谷伟玲董世奇樊慧蓉司端运
张爱杰,李 偲,刘羽康,张英华,谷伟玲,董世奇*,樊慧蓉*,谷 元,司端运
甘草苷体内暴露特征及体外跨膜转运机制研究
张爱杰1, 2,李 偲3,刘羽康3,张英华4,谷伟玲4,董世奇1, 2*,樊慧蓉1, 2*,谷 元5,司端运5
1. 中国医学科学院放射医学研究所,天津 300192 2. 中国医学科学院创新药物放射性药代研究重点实验室,天津 300192 3. 天津中医药大学,天津 301617 4. 吉林省中医药科学院,吉林 长春 132012 5. 天津药物研究院,天津 300462
总甘草素是甘草苷在体内的主要暴露形式,对两者在大鼠体内暴露特征及体外跨膜转运机制进行研究,为以甘草苷单体为新药的进一步开发提供依据。采用LC-MS/MS分析方法测定大鼠给药后不同时间点血浆样品中的总甘草素浓度,并应用WinNonlin.6.3软件采用非房室模型的统计矩法计算药代动力学参数;同时测定大鼠ig给药后组织匀浆中的浓度,考察不同类型甘草素在各组织中的暴露特征;应用体外MDCK-MDR1细胞模型,研究甘草苷、甘草素的跨膜转运能力及其机制。ig给药后在大鼠体内,甘草苷不呈线性动力学特征;血浆和大部分组织中主要以甘草素的II相结合产物存在,肝、子宫、卵巢、胃和肠组织中主要为游离甘草素;总甘草素暴露量排序为肠>血浆>肝>肾>肺>胃>子宫>卵巢>脂肪>心>脾>肌肉>睾丸,且不易产生组织蓄积现象;甘草素跨膜转运能力较甘草苷良好,且均不是P-gp转运体的底物。甘草苷不呈线性动力学吸收特征;甘草素在组织中暴露特征表现为不同组织中甘草素的存在形式和分布程度差异较大,总甘草素不易产生组织蓄积现象;两者均为被动扩散跨膜转运方式。
甘草苷;甘草素;暴露特征;吸收动力学;组织分布;跨膜转运机制
甘草为豆科植物甘草Fisch.、胀果甘草Bat. 及光果甘草L.的干燥根及根茎;其性味甘平,归心、肺、脾、胃经,具有补脾益气、清热解毒、祛痰止咳、缓急止痛、调和诸药的功效[1]。甘草的现代药理研究显示其具有抗炎、抗菌、保肝、抗氧化、抗癌、降糖、免疫调节等多种药理作用[2-3]。甘草的主要活性成分有三萜类、黄酮类、多糖类[4],具有广泛的生物活性。目前,甘草属中分离得到的黄酮类化合物及其衍生物有300多种[5],具有抗菌、抗病毒、保肝、抗肿瘤等药理作用。甘草苷是甘草中主要的黄酮类化合物,本课题组前期研究发现大鼠单次ig 30、100 mg/kg甘草苷后具有显著的抗垂体后叶素诱导的心肌缺血作用,表现为抑制心电图J点变化。因此,甘草苷拟作为抗心肌缺血Ⅰ类新药进行开发研究。
通过药物代谢和药动学研究药物的体内处置和动力学过程,将药物在血循环和靶器官的暴露水平与关键的临床表现(药效和不良反应)相关联,为了解动物或人体服用药物后的生物学和物理化学过程及机制提供科学依据[6]。已有文献报道提示,甘草素是甘草苷在体内的代谢产物之一,甘草苷进入体内主要是以甘草素的形式被吸收[7-10]。为了确定甘草苷经口给药后在体内的主要暴露形式,本研究首先进行了一系列预实验。结合药效学的给药方式和有效剂量,大鼠单次ig给予30 mg/kg甘草苷后大鼠血浆中检测到少量甘草苷(最高血药浓度约为50 ng/mL),而血浆中总甘草素平均最高浓度为(3.166±1.616)μg/mL,研究结果提示甘草苷原型不是甘草苷胃肠道给药后在体内的主要存在形式[10],同时血浆中总甘草素(包括游离甘草素、甘草素硫酸结合物、甘草素葡萄糖醛酸结合物)的暴露量远远大于游离甘草素的暴露量。吸收和分布是药物进入机体到达作用部位的重要过程,药物吸收的多少及快慢直接影响到药物在体内发挥的作用[11]。其中药物的吸收过程与转运密切相关,且甘草有“解百药毒”的特性[12],其机制之一为延缓和减少药物吸收,目前也有研究表明甘草这一特性与其跨膜转运功能有关[13]。基于此,本研究以含有葡萄糖醛酸酶和硫酸酯酶的H-2型水解酶,测定未经水解及酶水解后样品中甘草素的含量表征游离甘草素和总甘草素,定量分析两者在大鼠体内的吸收动力学、组织分布速度和程度及消除特点,总结归纳暴露特征,从而间接表征甘草苷在大鼠体内的动态变化;以甘草苷和甘草素为待测物分别评价其跨膜转运能力、探究跨膜转运机制等,为甘草苷的进一步研究开发提供参考数据。
1 材料
1.1 药品与试剂
甘草苷(批号20121211,质量分数为98.23%)、甘草素(批号20120401,质量分数为100%),吉林省中医药科学院;柚皮素(批号071100023,质量分数为99.2%),河南天方药业有限公司;羧甲基纤维素钠(分析纯)、-甲基-吡咯烷酮(分析纯)、甲酸(色谱纯)、甲酸铵(分析纯),天津市光复精细化工研究所;无水乙醇(分析纯),天津市化学试剂供销公司;甲醇(色谱纯)、冰乙酸(色谱纯),天津康科德科技有限公司;β-葡萄糖醛酸酶(Type H-2 From Helix Pomatia,批号011M7405),SIGMA-ALDRICH公司;磷酸二氢铵(分析纯),天津市化学试剂三厂;肝素钠注射液(批号20111006),天津市生物化学制药厂;DMEM培养基(批号NYM1045),美国Hyclone公司,使用前4 ℃保存;胎牛血清(批号1227693),美国Gibco公司,使用前−25 ℃保存;Trypsin-EDTA(批号1155732),美国Gibco公司,使用前4 ℃保存;超纯水,实验室自制。
1.2 实验动物
SPF级健康Wistar大鼠,雌雄各半,7~10周龄,体质量200~300 g。北京市维通利华实验动物科技有限公司提供,生产单位许可证编号:SCXK(京)2012-0001,动物质量合格证编号:No.11400700015112、No.11400700018465。本研究的动物使用方案已经获得天津市新药安全评价研究中心IACUC批准,IACUC号为2013071901。
1.3 仪器
液相色谱系统(配备LC-20AD型输液泵、DGU-20A3型脱气机、CTO-20A型柱温箱、SI L-20A型自动进样器、CBM-20A系统控制),日本Shimadzu公司;Diamonsil®C18色谱柱(150 mm×4.6 mm,5 µm),Dikma科技公司;质谱系统(配备API 4000 Q-TRAP质谱仪,ESI(电喷雾离子化)离子化源和Analyst 1.5.2分析数据处理系统工作站),美国Applied Biosystems公司;17R台式高速冷冻离心机,美国Thermo Scientific公司;Turbo Vap LV型样品浓缩仪,美国Caliper公司;700E型全自动均质器,美国TOMTEC公司;BM-40型纯水制备系统,北京中盛茂源科技发展有限公司;Eppendorf手动单道加样器,德国Eppendorf公司;ZHWY-110X50型恒温水浴震荡仪,上海智城分析仪器制造有限公司;IMS-40全自动雪花制冰机制冰机,常熟市雪科电器有限公司;Olympus- CKX41倒置显微镜,日本Olympus公司;二氧化碳培养箱,美国Thermo公司;Millcell ERS-2跨上皮电阻仪,美国Millipore公司;12孔聚碳酸酯膜转运(Transwell®),美国Corning公司;单人超净工作台及生物安全柜,力康生物医疗有限科技公司。
2 方法
2.1 LC-MS/MS条件
2.1.1 色谱条件 色谱柱为Diamonsil®C18(150 mm×4.6 mm,5 µm);柱温40 ℃;流动相为甲醇(A)-0.5 mmol/L甲酸铵溶液(含0.2%甲酸和10%甲醇,B)(52.5∶47.5);体积流量0.6 mL/min;进样体积为3 µL;运行时间为10 min(5~9.8 min切换进入质谱)。
2.1.2 质谱条件 ESI电喷雾离子源,离子喷雾电压−3000 V;温度500 ℃;喷雾气344.74 kPa(50 psi);加热气551.60 kPa(80 psi);卷帘气68.95 kPa(10 psi);碰撞气55.16 kPa(8 psi);负离子方式检测,扫描模式为多反应监测(MRM);甘草素碰撞能量为−40 V,内标物柚皮素碰撞能量为−25 V;甘草素/为255.0~119.0;内标物柚皮素/为271.0~151.0。
2.2 吸收动力学
2.2.1 给药溶液的配制 单次ig给药:准确称量甘草苷120、240、480 mg,在乳钵中研磨状态下缓缓加入0.5%羧甲基纤维素钠20 mL,分别制成6、12、24 mg/mL的药液。iv给药:准确称量甘草苷150 mg,在乳钵中研磨状态下缓缓加入2 mL-甲基吡咯烷酮,再加1 mL无水乙醇,完全溶解后,加入7 mL生理盐水,混匀,制成15 mg/mL的药液。
2.2.2 动物给药 单次ig给药:健康Wistar大鼠18只,体质量(245±25)g,雌雄各半,给药前禁食16 h,实验期间自由进食与饮水。按30、60、120 mg/kg的剂量单次ig给药,给药容积为5 mL/kg,每个剂量组6只。iv给药:健康Wistar大鼠6只,雌雄各半,体质量(272±40)g,给药前不禁食,试验期间自由进食与饮水。按30 mg/kg的剂量经尾iv,给药容积为2 mL/kg。
2.2.3 血浆样品的采集 单次ig给药:单次ig给药后0.167、0.5、1、2、3、4、6、9、12、17、24、30 h,分别自眼眶后静脉丛采血200 µL,肝素抗凝, 4 ℃,12 000 r/min,离心10 min,分离血浆,分装于EP试管中,−80 ℃冷冻保存。iv给药:iv给药后0.083、0.25、0.5、1、2、3、4、6、9、12、17、24和30 h,分别自眼眶后静脉丛采血200 µL,肝素抗凝, 4 ℃,12 000 r/min,离心10 min,分离血浆,分装于EP试管中,−80 ℃冷冻保存。
2.2.4 血浆样品处理与分析 精密吸取50 µL大鼠给药后的血浆样品,置于已加入10 µL乙酸工作液(0.56 mol/L)的玻璃管中,轻摇混匀,加入25 µL β-葡萄糖醛酸酶溶液(100 U),涡旋30 s混匀,置于37 ℃恒温水浴1 h;从水浴中取出后,加入内标柚皮素溶液(2 µg/mL,甲醇配制)50 µL,涡旋30 s混匀,再加入2.5 mL乙酸乙酯,涡旋2 min,3000 r/min离心10 min(4 ℃);取上清液2 mL,40 ℃下氮气吹干,用100 µL 50%甲醇水溶液复溶,12 000 r/min离心5 min(4 ℃),上清进样3 µL,进行LC-MS/MS分析。
2.2.5 药动学参数计算 采用WinNonlin 6.3软件的非房室模型统计矩法计算药动学参数,其中血药浓度-时间曲线下面积(AUC0-t、AUC0-∞)采用梯形法计算,消除半衰期(1/2)采用Best Fit法计算,5 min时药物浓度(5min)、峰浓度(max)、达峰时间(max)采用实测值。
2.3 组织分布研究
组织分布实验采用ig给药方式(与药效学实验一致),研究甘草苷的分布特征。
2.3.1 给药溶液的配制 精密称取甘草苷300 mg,加入0.5% 羧甲基纤维素钠25 mL研匀至混悬,临用现配,即得质量浓度为12 mg/mL的ig药液。
2.3.2 动物给药 健康Wistar大鼠18只,雌雄各半,体质量180~238 g。进行3个时间点(2、6、17 h)的组织分布实验,每个时间点6只,雌雄各半。给药前大鼠禁食16 h,给药后4 h进食,实验期间自由饮水。按60 mg/kg的剂量ig给药,给药容积为5 mL/kg。
2.3.3 生物样品的采集 分别于给药后2、6、17 h乙醚吸入麻醉手术剪断颈动脉取血至肝素化试管中,于10 min内置于离心机中离心(4℃、3000 r/min,离心5 min),分装于EP试管中,−80℃冷冻保存;取血后乙醚过量麻醉脱颈处死动物,分别剖取脑、肌肉、脂肪、睾丸、卵巢、子宫、肝、脾、肾、肺、心、胃、小肠,纯水冲洗,滤纸吸干水分,称定质量,剪刀剪碎,放入EP试管中。将EP试管置于全自动匀浆机中,按照5 mL/g对应体积加入纯水进行匀浆,将各组织匀浆置于−80℃冷冻保存,待测。
2.3.4 组织样品处理与分析 除脏器组织样品以大鼠各组织匀浆代替血浆外,其他实验操作同“2.2.4”项。上述处理过程用于组织样品中总甘草素浓度的测定。应用于游离甘草素的浓度测定时,不加入乙酸工作液和β-葡萄糖醛酸酶进行孵育,其余处理过程与总甘草素的浓度测定一致。
2.3.5 数据处理
(1)组织含量计算:由LC-MS/MS法测定并经Analyst1.5.2数据处理软件计算获得的各组织样品的质量浓度单位为ng/mL,换算成各组织含量(ng/g)需乘以匀浆体积与各组织质量的比值(5 mL/g),具体公式如下。
组织含量=组织匀浆测定浓度×5
(2)组织暴露量计算:采用梯形法计算总甘草素和游离甘草素在各组织中的AUC0-17 h,以考察药物在各组织中的分布程度。
2.4 跨膜转运机制研究
2.4.1 无血清细胞培养基-多药耐药蛋白1(MDCK-MDR1)细胞模型的建立 Madin-Darby canine kidney(MDCK)-MDR1细胞是将人类的MDR1基因转染到MDCK细胞上产生的[14-15],多药耐药蛋白(MDR1)也称为P-糖蛋白(P-gp)[16]。MDCK-MDR1细胞广泛应用于P-gp底物和抑制剂的体外快速筛选研究。该细胞株由日本富士生物医药研究所友情赠送。细胞培养方法如下[17]:MDCK-mock和MDCK-MDR1细胞于常规培养皿内,以高糖DMEM培养液(含10% FBS)在37 ℃、5% CO2的培养箱内培养,用含0.25% EDTA的胰酶消化,用新鲜培养液调细胞密度至2×105/mL接种于Transwell聚碳酸酯膜12孔板中(膜面积为1.13 cm2)。在顶端侧(AP)每孔加0.5 mL细胞悬液,基底侧(BL)每孔加1.5 mL新鲜培养基,每天换液,连续培养6 d,得到完全分化的细胞单层。
本实验所用MDCK-mock和MDCK-MDR1细胞均为25~30代。
2.4.2 MDCK-mock和MDCK-MDR1单层细胞电阻(TEER)测定 MDCK-mock和MDCK-MDR1细胞接种于Transwell膜上培养6 d;将Millcell ERS-2电阻仪的电极放入含有Hank's平衡盐溶液(HBSS)的烧杯中,预平衡20 min;移去培养板中的培养液,AP侧每孔加预热的HBSS 0.5 mL,BL侧每孔加预热的HBSS 1.5 mL,37 ℃平衡20 min,洗去细胞表面的杂质;移走HBSS,重新加入预热的HBSS,测定跨膜电阻值;用1个空白载体重复上述步骤以获得空白值。
2.4.3 细胞单层的双向转运实验[17]
(1)AP→BL方向的转运试验:吸去Transwell小室AP和BL端的培养基,加入37 ℃预热的HBSS溶液,37 ℃平衡20 min;吸去HBSS,在AP端加入0.5 mL预热的含有甘草苷/甘草素的HBSS溶液,BL端加入1.5 mL空白HBSS溶液;37 ℃恒温振荡器培养1.0 h,振荡器转速为80 r/min;吸取BL端的转运液,测定转运液中的甘草苷/甘草素浓度。
(2)BL→AP方向的转运试验:吸去Transwell小室AP和BL端的培养基,加入37 ℃预热的HBSS溶液,37 ℃平衡20 min;吸去HBSS,在AP端加入0.5 mL空白HBSS溶液,BL端加入1.5 mL预热的含有甘草苷/甘草素的HBSS溶液;37 ℃恒温振荡器培养1.0 h,振荡器转速为80 r/min;吸取AP端的转运液,测定转运液中的甘草苷/甘草素浓度。
2.4.4 数据的处理与分析
(1)TEER的计算:采用TEER值表征细胞间紧密连接的完整性,并按照公式计算。
TEER=(测定电阻值-空白值)×单层表面积
(2)表观渗透系数(app)的计算:采用app值的大小表征药物透过单层细胞的能力和药物吸收的速度以及程度。研究表明药物在Caco-2细胞模型的app与药物人体口服吸收程度相关性良好,且与药物在MDCK细胞和Caco-2细胞中的渗透性有良好相关性。
app=d/d×1/×1/0
d为药物在d时间内的透过量,为膜的表面积,0为初始浓度。
(3)外排率(E)的计算E值的大小反映药物外排的能力,通过E值可以推测药物是否为P-糖蛋白的底物。当待测药物的E≥2时,表明药物可能为肠道外排转运体的底物,当E≤0.5时,表示该药物可能为肠道摄入型转运蛋白的底物,需要做进一步研究。
E=app(BL-AP)/app(AP-BL)
3 结果
3.1 吸收动力学
3.1.1 大鼠单次ig给药的血浆总甘草素浓度-时间曲线 大鼠ig给药后,3个剂量组动物不同时间平均血浆总甘草素浓度-时间曲线比较见图1。
3.1.2 大鼠iv给药的血浆总甘草素浓度-时间数据 在30 mg/kg剂量下大鼠ig与iv给予甘草苷后,不同时间相应的平均血浆总甘草素浓度-时间曲线比较见图2。
3.1.3 大鼠单次ig、iv后血浆总甘草素药动学参数比较 大鼠单次ig、iv后血浆总甘草素药动学参数见表1。在大鼠单次ig甘草苷后,在30~120 mg/kg剂量内,与体内暴露量密切相关的药动学参数AUC0-30 h、AUC0-∞、max随剂量增加而增加,增加的比例小于剂量比;与分布、消除过程密切相关的1/2、清除率(CL)、分布表观容积(d)等参数与给药剂量不相关,不随给药剂量增加而呈明显变化。上述情况均表明大鼠单次ig甘草苷后,在30~120 mg/kg剂量内不呈线性动力学特征。根据大鼠ig给予甘草苷后总甘草素的AUC0-30 h均值与同剂量下iv给予甘草苷后总甘草素的AUC0-30 h均值进行计算,大鼠ig甘草苷后的绝对生物利用度为80.6%。
3.2 组织分布研究
3.2.1 甘草苷的组织分布特点 大鼠ig 60 mg/kg甘草苷后在2、6、17 h这3个时间点平均总甘草素和游离甘草素在各组织/血浆的分布见表2。对各组织/血浆中总甘草素和游离甘草素含量进行分析比较,结果表明:(1)大鼠ig给予甘草苷后,血浆中2 h和17 h时间点检测不到游离甘草素,6 h时游离甘草素也非常少,而水解后的总甘草素浓度较高,表明大鼠血浆主要以甘草素的II相结合产物形式存在。(2)大鼠ig给予甘草苷后,各组织中甘草素分布的存在形式和组织暴露量不同,大鼠血浆主要以甘草素的II相结合产物形式存在;脾、子宫、卵巢、肝、胃和肠组织中直接测得游离甘草素的含量与组织样品经酶水解后测得总甘草素含量相似,提示上述组织中游离甘草素为主要暴露物质;其余组织结合型甘草素与游离甘草素共存。ig给药后6 h药效靶器官心组织中总甘草素的含量较游离甘草素高1倍,至给药后17 h总甘草素与游离甘草素含量相似,且游离甘草素含量下降缓慢,表明结合型甘草素在心组织中可能逐渐全部转化为游离甘草素。(3)除胃中甘草素的含量在给予甘草苷2 h后最高外,其余组织均在6 h达到分布峰值,之后随着时间的延长呈迅速下降趋势。大鼠ig给药17 h,即约3个半衰期后,大部分组织中已检测不到甘草素或含量已降至分布峰值的1/10~1/20,表明不会产生组织蓄积趋势。
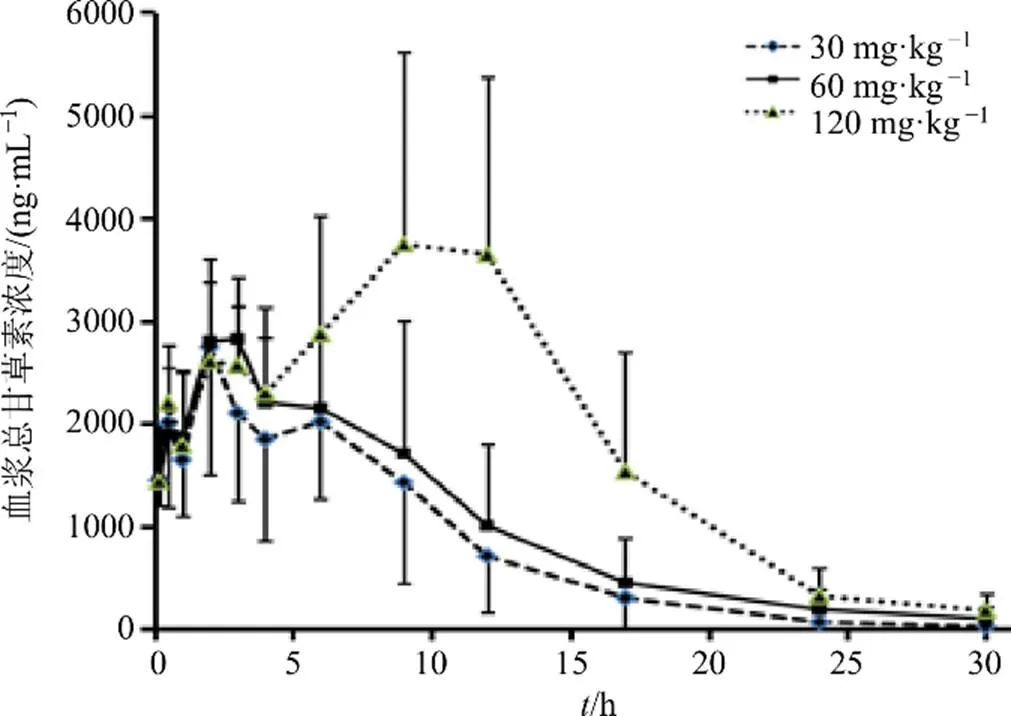
图1 大鼠ig给予3剂量甘草苷后的平均血浆总甘草素浓度- 时间曲线(n= 6)
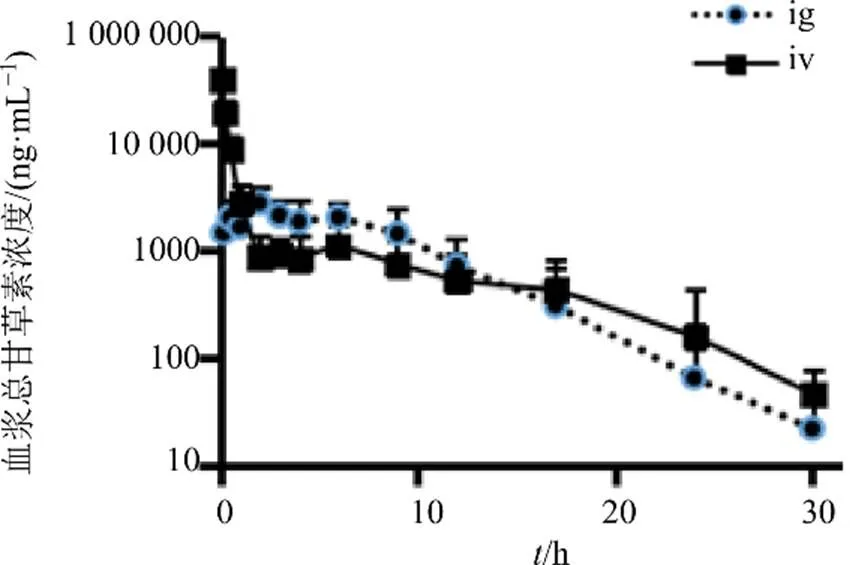
图2 大鼠ig与iv给予30 mg/kg甘草苷后平均血浆总甘草素浓度-时间曲线比较 (半对数坐标,n= 6)
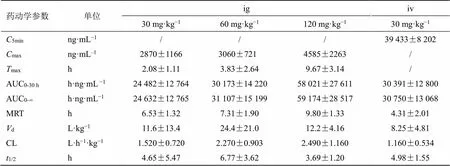
表1 大鼠单次ig、iv后总甘草素药动学参数比较(n= 6)
*= 3
3.2.2 甘草苷的组织分布程度 按“2.3.5”项下方法计算的各组织/血浆中总甘草素和游离甘草素的分布程度AUC0-17 h和游离甘草素占总甘草素的比例(AUCF/AUCT)结果见表3。
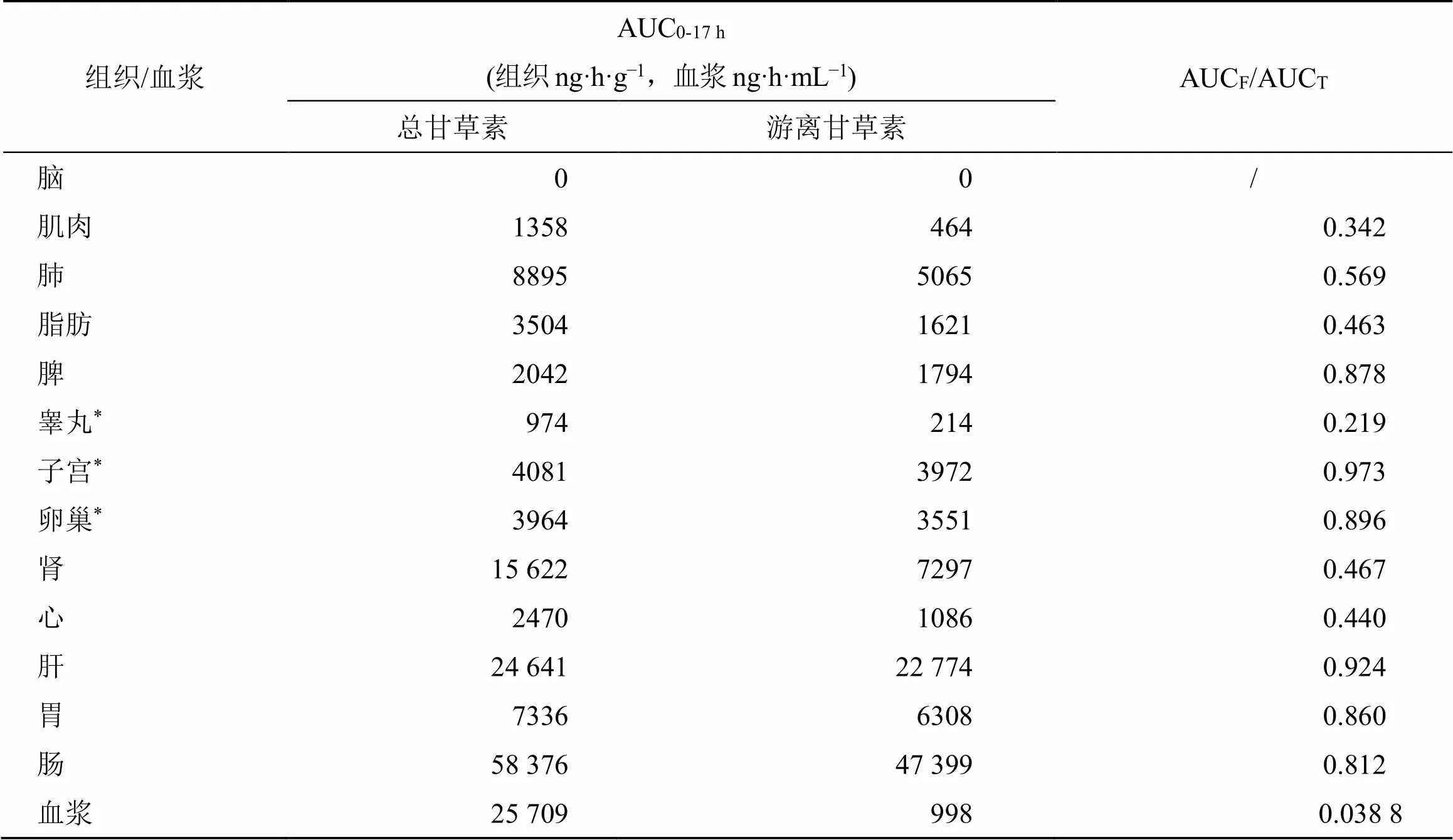
表3 大鼠ig给予甘草苷后各组织和血浆中总甘草素和游离甘草素的AUC0-17 h比较
AUCF为游离甘草素的AUC0-17 h;AUCT为总甘草素的AUC0-17 h;*= 3
AUCFis the value of AUC0-17 hfor free liquiritigenin; AUCTis the value of AUC0-17 hfor total aglycone liquiritigenin;*= 3
根据各组织中总甘草素和游离甘草素的组织暴露量AUC0-17 h结果可知,总甘草素分布程度肠>血浆>肝>肾>肺>胃>子宫>卵巢>脂肪>心>脾>肌肉>睾丸;游离甘草素分布程度:肠>肝>肾>胃>肺>子宫>卵巢>脾>脂肪>心>血浆>肌肉>睾丸。总甘草素在肠、肝、肾、胃、肺分布相对较高;在雌性生殖器官卵巢、子宫中的分布相对高于雄性生殖器官睾丸;在心、脾、脂肪、肌肉中分布相对较少;无论结合型的还是游离型甘草素均不能到达脑组织,因此脑中检测不到甘草素。
3.2.3 总甘草素的组织含量/血浆浓度比 各时间点组织样品中总甘草素平均含量与同时间点血浆的平均浓度比值见表4。
肺、肝的总甘草素组织/血浆比随时间变化基本维持一致;肌肉、脾、卵巢、肾、心、胃和肠的总甘草素组织/血浆比随时间推进迅速增加,结果表明,除肺和肝外,总甘草素在其余组织的消除过程和血浆的消除过程不一致。
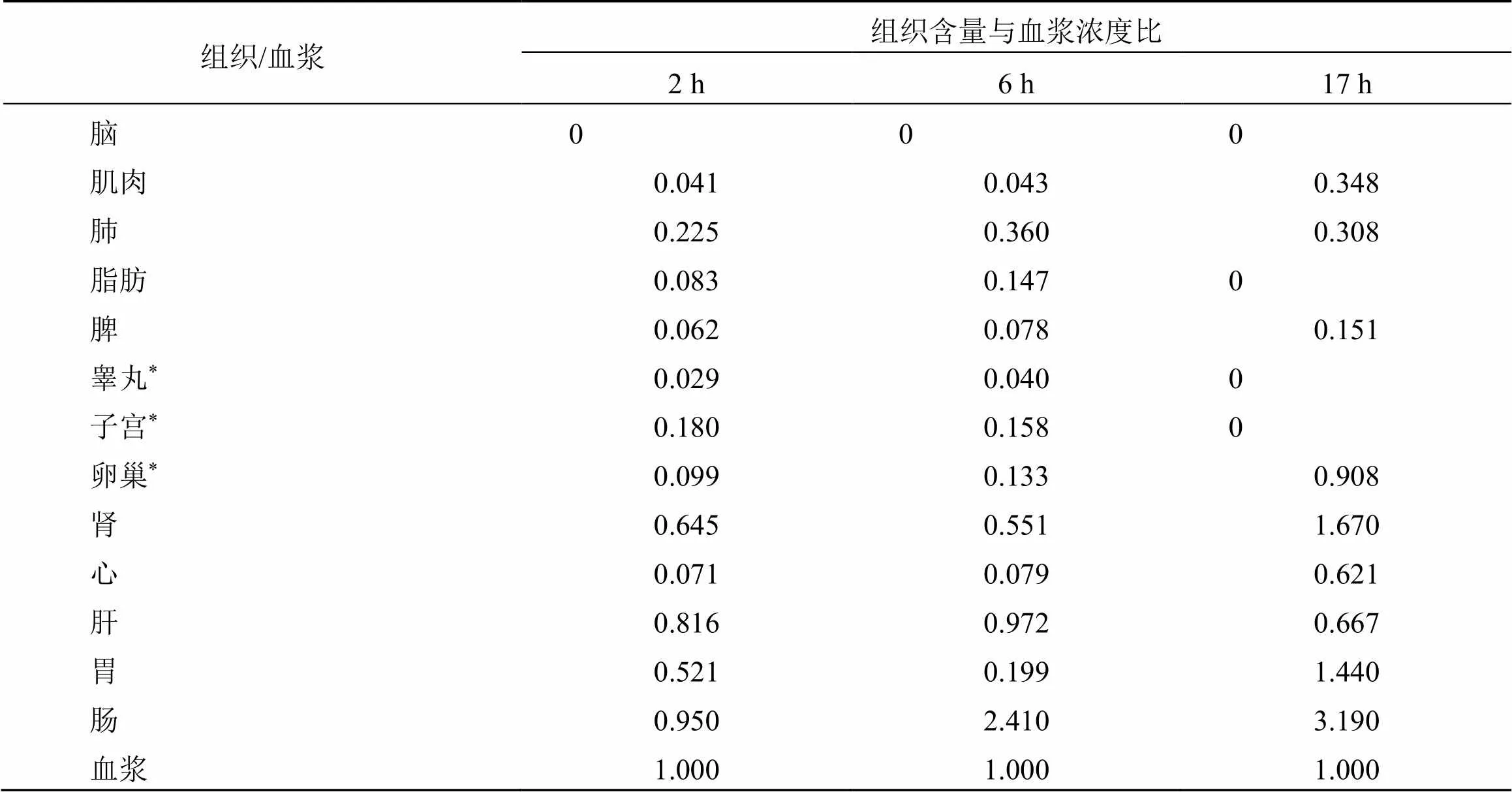
表4 组织样品中总甘草素平均含量与同时间点的平均血浆浓度比值(n= 6)
*= 3
3.3 跨膜转运机制研究
3.3.1 MDCK-MDR1细胞模型的验证 MDCK- mock细胞单层的TEER值到第6天达到(245±13)Ω·cm2>150 Ω·cm2,MDCK-MDR1细胞单层的TEER值到第6天达到(242±10)Ω·cm2>150 Ω·cm2,表明MDCK-mock和MDCK-MDR1细胞单层的致密性与完整性均良好,符合实验要求。
3.3.2 甘草苷和甘草素在MDCK-MDR1细胞的双向转运 MDCK-MDR1细胞模型,甘草苷5 μmol/L给药,甘草素10 μmol/L给药,得到的双向转运app和E值,见表5。
本研究中甘草苷在MDCK-MDR1细胞模型摄入方向的app约为2×10−6cm/s,说明甘草苷跨膜转运能力中等;甘草素的app约为17.0×10−6cm/s,说明甘草素跨膜转运能力良好。甘草苷和甘草素在MDCK-MDR1细胞模型中E<2,提示甘草苷和甘草素主要以被动扩散形式跨膜转运,均不存在P-糖蛋白外排转运蛋白介导的跨膜转运机制。

表5 甘草苷和甘草素在MDCK-MDR1细胞模型的Papp值和RE
4 讨论
本研究根据文献报道的黄酮类化合物吸收和代谢特点[18],针对性地对大鼠体内甘草苷、总甘草素和游离甘草素暴露量进行预实验研究,研究结果表明甘草苷在血浆中的主要存在形式为甘草素的II相结合产物,因此本实验采取β-葡萄糖醛酸酶水解总苷的方式定量分析总甘草素和游离甘草在体内吸收分布中的暴露特征从而间接表征甘草苷在体内的动态变化。另外,采用MDCK-MDR1细胞模型研究甘草苷和甘草素各自的跨膜转运能力和转运方式,为进一步阐明药物吸收机制提供了依据。
本实验中甘草苷在大鼠体内的吸收不呈线性动力学特征,这需要进一步阐明其饱和机制,再根据非线性动力学的规律,指导临床试验的用药方案和剂量调整。药物不呈现线性动力学特征的主要原因是体内的某些系统达到饱和,如酶和转运体、血浆蛋白结合的饱和、肾小管重吸收的饱和等[19]。在当前甘草苷临床前药动学研究中,大鼠的药动学参数AUC0-t、max随剂量增加而增加,但增加的比例均小于剂量比,说明体内可能存在某种饱和机制,但机制的阐明还需进一步的试验,这种非比例化剂量反应关系给预测人体安全有效的剂量范围带来复杂性[19]。此外,本实验发现血浆中总甘草素的药-时曲线存在双峰现象,进一步研究了甘草苷(60 mg/kg)单次ig给药后胆汁排泄情况,结果发现胆汁中总甘草素0~48 h累积排泄分数为(32.1±10.7)%,提示ig给药后总甘草素存在肝肠循环现象,可能是药时曲线出现双峰的原因。
在组织分布中,相对于其他组织,心脏中的游离甘草素浓度在6 h和17 h差别不大,而其他组织17 h时的游离甘草素的浓度比6 h显著降低,推测心脏组织中的甘草素消除较慢,从现有数据分析这一结果可能会导致两方面作用,一是延长药效作用,二是可能会引起心脏组织的蓄积。前期预实验采集给药后24 h组织样本并进行测定,结果表明大部分组织已检测不到甘草素,其中有一半的大鼠心脏组织中检测不到甘草素,排除个体差异的因素,这一结果表明甘草素在体内24 h即可大部分消除,不会引起心脏组织的蓄积。因此,心脏组织比较其他组织消除较慢这一现象可能会使药物作用延长,这一点应该是支持其心血管系统临床应用的依据。甘草苷拟作为抗心肌缺血类I类新药进行开发研究,理论上,心脏是甘草苷(甘草素)发挥其抗心肌缺血作用的靶器官之一,但绝不是唯一发挥药效作用的关键因素,单纯地从药物在心脏组织的分布量来衡量其心血管系统作用是不全面的,其更全面深刻的作用机制还需进行进一步的试验研究。
药物吸收、分布、排泄仅是发生空间位置上的迁移,统称为转运[20]。药物的体内动态就是其在体内一系列跨膜转运的综合效果。转运体的初步研究表明甘草苷和甘草素主要以被动扩散形式跨膜转运,均不存在P-gp外排转运蛋白介导的跨膜转运机制。因此,不需关注基于P-gp的药物-药物相互作用,而且不呈线性动力学的原因也可基本排除这一因素。大多数药物对于机体来说是外源性物质,且具有一定的脂溶性,推测甘草苷和甘草素的跨膜转运机制主要是被动扩散中的溶解扩散[21-22],是否为通过膜上含水孔道转运的限制扩散还需更进一步的研究。药物的跨膜转运是相当复杂的过程,药物的跨膜转运不仅直接参与药物的吸收、分布、排泄等药动学行为,同时与分布于靶器官的有效药物浓度密切相关,因此明确药物的跨膜转运能力、机制以及对药物体内动力学的调控作用[23],对于新药研发和评价意义重大。
利益冲突 所有作者均声明不存在利益冲突
[1] 中国药典 [S]. 一部. 2010: 80-81.
[2] Asl M N, Hosseinzadeh H. Review of pharmacological effects ofsp. and its bioactive compounds [J]., 2008, 22(6): 709-724.
[3] 任玲. 甘草有效成分的药理活性研究 [J]. 生物技术世界, 2016, 13(5): 227.
[4] 乔湜, 李清艳, 孙瑞芳, 等. 复方甘草口服溶液中7种成分的UPLC-MS/MS分段离子切换法测定 [J]. 中国医药工业杂志, 2014, 45(5): 470-473.
[5] 黄雨婷, 迟宗良, 王姝梅, 等. 甘草中的黄酮类成分及其抗肿瘤活性研究进展 [J]. 中国新药杂志, 2017, 26(13): 1532-1537.
[6] 李桦, 庄笑梅. 药代动力学人体预测及其在新药研发中的应用 [J]. 中国药理学与毒理学杂志, 2013, 27(4): 611-615.
[7] Kitagawa I, Chen W Z, Taniyama T,. Quantitative determination of constituents in various licorice roots by means of high performance liquid chromatography [J]., 1998, 118(11): 519-528.
[8] Zuo F, Zhou Z M, Yan M Z,. Metabolism of constituents in Huangqin-tang, a prescription in traditional Chinese medicine, by human intestinal flora [J]., 2002, 25(5): 558-563.
[9] Dong S Q, Fan H R, Li Q S,. LC-MS/MS method for quantification of liquiritigenin in rat plasma: Application to pharmacokinetic study of liquiritin [J]., 2016, 8(1): 53-60.
[10] 董世奇, 樊慧蓉, 李全胜, 等. 甘草苷在大鼠体内的代谢途径研究 [J]. 中草药, 2014, 45(17): 2499-2505.
[11] 李志华, 颜苗, 张毕奎, 等. 基于药动学的甘草配伍减毒机制研究进展 [J]. 中草药, 2015, 46(23): 3611-3616.
[12] 覃一枫, 魏崴, 杭晓敏, 等. 基于GC-MS技术的甘草总黄酮对伊立替康致肠炎小鼠的血浆代谢组学的影响研究 [J]. 中草药, 2018, 49(24): 5836-5842.
[13] 何丹, 颜苗, 李焕德, 等. 甘草提取物及其主要成分对Caco-2细胞膜上P-gp功能和表达的影响 [J]. 中国药学杂志, 2010, 45(10): 751-755.
[14] Pastan I, Gottesman M M, Ueda K,. A retrovirus carrying an MDR1 cDNA confers multidrug resistance and polarized expression of P-glycoprotein in MDCK cells [J]., 1988, 85(12): 4486-4490.
[15] Horio M, Chin K V, Currier S J,. Transepithelial transport of drugs by the multidrug transporter in cultured Madin-Darby canine kidney cell epithelia [J]., 1989, 264(25): 14880-14884.
[16] 胡亚, 刘运锋, 朱卫丰, 等. 马钱子生物碱类成分在MDCK-MDR1单层细胞模型中的转运机制研究 [J]. 中草药, 2019, 50(12): 2876-2883.
[17] 慈小燕, 夏媛媛, 曾勇, 等. 应用MDCK-MDR1细胞模型研究3-乙酰基-11-羰基-b-乙酰乳香酸经血脑屏障的通透性 [J]. 中国新药杂志, 2013, 22(10): 1125-1129.
[18] 周乐, 赵晓莉, 狄留庆, 等. 黄酮类化合物口服吸收与代谢特征及其规律分析 [J]. 中草药, 2013, 44(16): 2313-2320.
[19] 张志伟, 李正, 庄笑梅. 小分子药物非线性药代动力学特征、机制与预测评估研究进展 [J]. 国际药学研究杂志, 2018, 45(11): 805-812.
[20] 伊秀林, 司端运, 刘昌孝. 应用药物转运体的药代动力学评价 [J]. 药物评价研究, 2010, 33(5): 341-346.
[21] Dobson P D, Kell D B. Carrier-mediated cellular uptake of pharmaceutical drugs: An exception or the rule? [J]., 2008, 7(3): 205-220.
[22] Sugano K, Kansy M, Artursson P,. Coexistence of passive and carrier-mediated processes in drug transport [J]., 2010, 9(8): 597-614.
[23] 慈小燕, 崔涛, 武卫党, 等. 药物的跨膜转运机制研究进展 [J]. 药物评价研究, 2018, 41(6): 973-979.
exposure characteristic andtransport mechanism of liquiritin
ZHANG Ai-jie1, 2, LI Cai3, LIU Yu-kang3, ZHANG Ying-hua4, GU Wei-ling4, DONG Shi-qi1,2, FAN Hui-rong1, 2, GU Yuan5, SI Duan-yun5
1. Institute of Radiation Medicine, Chinese Academy of Medical Sciences, Tianjin 300192, China 2. Key Laboratory of Radiopharmacokinetics for Innovative Drugs, Chinese Academy of Medical Sciences, and Institute of Radiation Medicine, Tianjin 300192, China 3. Tianjin University of Traditional Chinese Medicine, Tianjin 301617, China 4. Jilin Academy of Chinese Medicine Sciences, Changchun 132012, China 5. Tianjin Institute of Pharmaceutical Research, Tianjin 300462, China
The total aglycone liquiritigenin represents the primary exposure of liquiritinin rats. The objective of this study was to investigate the characteristics of exposure and transport mechanism of liquiritinin rats and. The results could provide the evidence of liquiritin for its further development as a new drug.The concentration of total aglycone liquiritigenin at different timing points in rat plasma and tissue distribution samples was determined by LC-MS/MS method after an oral administration of liquiritin in rats. The pharmacokinetic parameters were calculated by WinNonlin software using Rosenbluth method of non-compartment model. The exposure of total aglycone liquiritigenin and free liquiritigenin in rat tissues was investigated. And the transport mechanism of liquiritin and liquiritigenin was clarified using the MDCK-MDR1 cells.Liquiritin did not undergo linear pharmacokinetic characteristic after a single oral dose in rats. The phase II conjugation metabolites of liquiritigenin were the major exposure in plasma and most rat tissues. However, free liquiritigenin was primarily distributed in the tissue of rat liver, uterus, ovary, stomach and intestine. The exposure of total aglycone liquiritigenin in rat tissues did not show any accumulation trends. And the sequence of exposure was: intestine>plasma>liver>kidney>lung>stomach>uterus>ovary>fat>heart>spleen>muscle>testicle.The transport results indicated that both liquiritin and liquiritigenin were not the substrate of P-gp. And the transcellular transport of liquiritigenin from apical to basolateral membrane was higher than that of liquiritin in MDCK-MDR1 cells.Liquiritin did not show linear pharmacokinetic characteristic after a single oral dose in rats. The characteristics of exposure of liquiritigenin in rat tissues indicated that the extent of distribution and type of liquiritigenin were different among tissues. The exposure of total aglycone liquiritigenin in rat tissues did not show any accumulation trends. The transcellular transport of liquiritin and liquiritigenin was mediated by passive diffusion.
liquiritin, liquiritigenin, exposure characteristic, absorptionkinetics, tissue distribution, transcellular transport mechanism
R283
A
0253 - 2670(2021)07 - 2053 - 09
10.7501/j.issn.0253-2670.2021.07.022
2020-05-28
国家重点研发计划项目(2018YFC1708203);中国医学科学院中央级公益性科研院所基本科研业务费专项资金资助(2018PT35031);内蒙古自治区科技重大专项(2019ZD004);天津市自然科学基金青年基金项目(20JCQNJC00320)
张爱杰,男,助理研究员,研究方向为药动学。E-mail: zhangaijie1986@163.com
董世奇,男,博士,研究方向为药动学。E-mail: dongsq1314@126.com
樊慧蓉,女,研究员,硕士导师,研究方向为药动学。E-mail: fanhr99@163.com
[责任编辑 王文倩]
