AlCl3和GaCl3上AsH3催化氧化反应机理的理论研究
2021-04-07杨春晓张艳琨张可欣李粉吉杜晓琴李相华夏福婷
杨春晓,张艳琨,张可欣,李粉吉,杜晓琴,李相华,夏福婷
(云南民族大学 云南省高校民族地区资源清洁转化重点实验室,云南 昆明 650500)
当前国家极力提倡生态文明建设和工业绿色发展,因此工业尾气中CO生产化工产品受到了广泛关注[1-3].然而黄磷尾气和电石炉尾气因含有微量的AsH3等非常规污染物往往会导致后续碳一化工生产催化剂中毒,并影响产品品质[4-7].因此对黄磷尾气和电石炉尾气中AsH3的净化意义重大.目前已报到的AsH3净化方法主要包括化学吸收法、吸附法、燃烧法、催化氧化法等[8].前面几种方法存在易造成设备腐蚀、氧化砷难以全量捕集和造成二次污染等问题[9-14].催化氧化法是在催化剂作用下,将AsH3催化氧化转化为氧化砷产物[15]并储存在催化剂上.该方法极大减少了砷物种的暴露途径,大幅减少了二次污染,因此被认为是净化AsH3实施性最强的技术.
对于AsH3催化氧化净化,林亦龙[16-17]等制备了一系列对AsH3有较好去除性能的催化剂,发现Cu-Fe和Cu-Mn二元类水滑石催化剂体现出较强的AsH3去除性能.此外还制备了不同合成条件下的类水滑石化合物(HTLC)前体,结果表明CuZnAl类水滑石催化剂的合成条件对AsH3的去除率有显著影响.严书梯[18]对矿冶废气中的AsH3进行吸附脱除研究,表明通过Cu(NO3)2改性的Hp型分子筛对AsH3表现出了更好的吸附性能.尽管AsH3催化氧化净化在实验方面已经取得了一定进展,但是AsH3的剧毒性带来的实验过程高风险,实验条件苛刻和高运行成本限制了人们对其深入研究,因此国内外目前对AsH3污染消除的研究并不多见,但近年来关于砷污染消除的理论研究越来越多[19-21].Prayut[22]等从理论上研究了原始的和掺杂了过渡金属的碳纳米管(SWCNTs)对AsH3的吸附性能,结果表明掺杂的SWCNTs比原始SWCNTs更适合气体分子吸附和检测.Li[23]等利用密度泛函理论(DFT)研究了水分子对AsH3催化氧化AsH3+O2+nH2O(n=0~5)的作用,结果表明添加(H2O)n(n=1~5)对氧化过程有促进催化作用,与不添加水分子(16.44 kcal/mol)的反应相比,反应势垒能降低3.27~10.70 kcal/mol.
前期研究发现氯化物能有效除砷[24-26],本文采用密度泛函理论方法研究在催化剂AlCl3和GaCl3上AsH3的催化氧化反应机理.本研究将为AsH3催化氧化高效催化剂的研制起指导作用.
1 计算详情
反应体系的反应物(RC)、过渡态(TS)、中间体(INT)和产物(PC)的构型均采用密度泛函理论(DFT)[27-29]方法在Gaussian 03程序包中在B3LYP/6-31++G(d, p)水平上进行全优化.对各驻点的频率进行考察,全实频的是稳定点,有唯一虚频的是过渡态.通过内禀反应坐标(IRC)[30-31]计算,以确定与过渡态相关联的反应物以及产物,并对零点能量(ZPE)[32]进行校正.
2 结果与讨论
在砷化氢(AsH3)的直接氧化反应和以AlCl3、GaCl3为催化剂的催化氧化反应途径中,我们考虑了分子模型在气相中的优化.图1为O2单体、AsH3、AlCl3和GaCl3在B3LYP/6-31++G(d,p)水平下优化的几何构型,计算表明在B3LYP/6-31++G(d, p)水平下三重态的O2分子比单重态的O2分子更稳定,因此本文选择O2分子的三重态作为反应物.图1中O-O键的键长为 0.121 5 nm,As-H键的键长为 0.150 6 nm,Al-Cl键的键长为 0.208 7 nm,Ga-Cl键的键长为 0.2127 nm,Cl-Ga-Cl、Cl-Al-Cl的键角均为120.00°,H-As-H的键角为91.33°.

图1 O2、AsH3、AlCl3和GaCl3单体在B3LYP/6-31++G(d,p)水平下优化的几何构型
2.1 直接氧化反应
2.1.1 途径Ⅰ(AsH3+O2)
在途径 Ⅰ 中,AsH3与一分子氧气(O2)反应生成H3AsO2,此途径分两步完成,A-RC1→A-TS1-1→A-INT1-1→A-TS1-2→A-PC1.如图2所示,在反应的初始态(A-RC1)中,As-H3和O1-H3键长分别为 0.149 3 nm 和 0.253 3 nm,当H3原子进攻O1原子时,O2原子转移到AsH3的As原子上,得到了过渡态A-TS1-1.过渡态A-TS1-1中As-H3、H3-O1的键长分别为 0.154 3、0.174 0 nm,对应的唯一虚频为 -1 018.73 icm-1.通过振动模式以及过渡态A-TS1-1的内禀反应坐标(IRC)计算可知在A-TS1-1到A-INT1-1的过程中,As-H3从 0.154 3 伸长至 0.309 0 nm;O1-H3从 0.174 0 nm 缩短至 0.097 2 nm.表明在第一步中主要是As-H3键的断裂和O1-H3键的形成.在第二步中O1原子脱离O2原子转移到As原子上,得到了过渡态A-TS1-2,其唯一虚频是 -630.23 icm-1,As-O1、O1-O2键长分别为 0.216 2、0.203 5 nm.通过内禀反应坐标(IRC)计算可知在A-TS1-2到A-PC1的过程中,As-O1从 0.216 2 nm 缩短至 0.177 5 nm;O1-O2从 0.203 5 nm 伸长至 0.286 6 nm,对应着O1-O2键的断裂和As-O1键的形成.
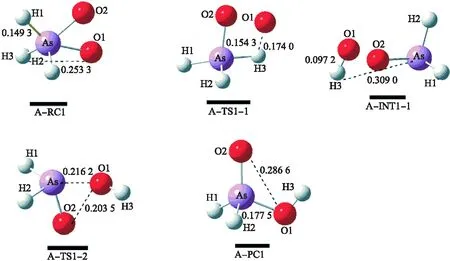
图2 AsH3-O2直接氧化反应的各驻点在B3LYP/6-31++G(d,p)水平下气相中优化的构型
2.1.2 途径Ⅱ((AsH3+2O2)
途径Ⅱ是在途径Ⅰ中的中间体A-INT1-1上增加1分子氧气(O2)的直接氧化反应,该途径分4步完成.如图3所示,在A-RC2中当H2原子进攻O3原子时,O4原子转移到As原子上,得到了过渡态A-TS2-1.过渡态A-TS2-1中As-H2和H2-O3的键长分别为 0.156 2 和 0.174 2 nm,对应的唯一虚频是 -971.57 icm-1,通过振动模式和过渡态A-TS2-1的内禀反应坐标(IRC)计算可知在A-TS2-1到A-INT2-1的过程中,As-H2从 0.156 2 伸长至 0.313 0 nm;H2-O3从 0.174 2 缩短至 0.097 2 nm.表明在第1步中主要是As-H2键的断裂和H2-O3键形成.随后,当O3原子脱离O4原子转移到砷化氢的As原子上,得到了过渡态A-TS2-2,其中As-O3和O3-O4键长分别为 0.209 6 和 0.201 8 nm,对应的唯一虚频是 -531.33 icm-1,通过振动模式和过渡态A-TS2-2的内禀反应坐标(IRC)计算可知在A-TS2-2到A-INT2-2的过程中,As-O3从 0.209 6 缩短至 0.175 9 nm;O3-O4从 0.201 8 伸长至 0.286 0 nm,这一步中主要是O3-O4键的断裂和As-O3键的形成.该途径的第3步为H1原子脱离As原子转移到O4原子上,得到了过渡态A-TS2-3,虚频为 -1 399.61 icm-1,As-H1和O4-H1的键长分别为 0.155 4 和 0.162 0 nm.从A-TS2-3到A-INT2-3的过程中,As-H1从 0.155 4 伸长至 0.234 6 nm ,O4-H1从 0.162 0 缩短至 0.096 8 nm,对应着As-H1键的断裂和O4-H1键形成.最后一步为O1原子脱离O2原子转移到As原子上得到过渡态A-TS2-4.其中As-O1和O1-O2键长分别为 0.202 4 和 0.204 2 nm,对应的唯一虚频是 -550.08 icm-1,通过振动模式和计算过渡态A-TS2-4的内禀反应坐标(IRC)可知从A-TS2-4到A-PC2的过程中,As-O1从 0.202 4 缩短至 0.175 6 nm ;O1-O2从 0.204 2 伸长至 0.294 3 nm,对应着O1-O2的断裂和As-O1键形成.
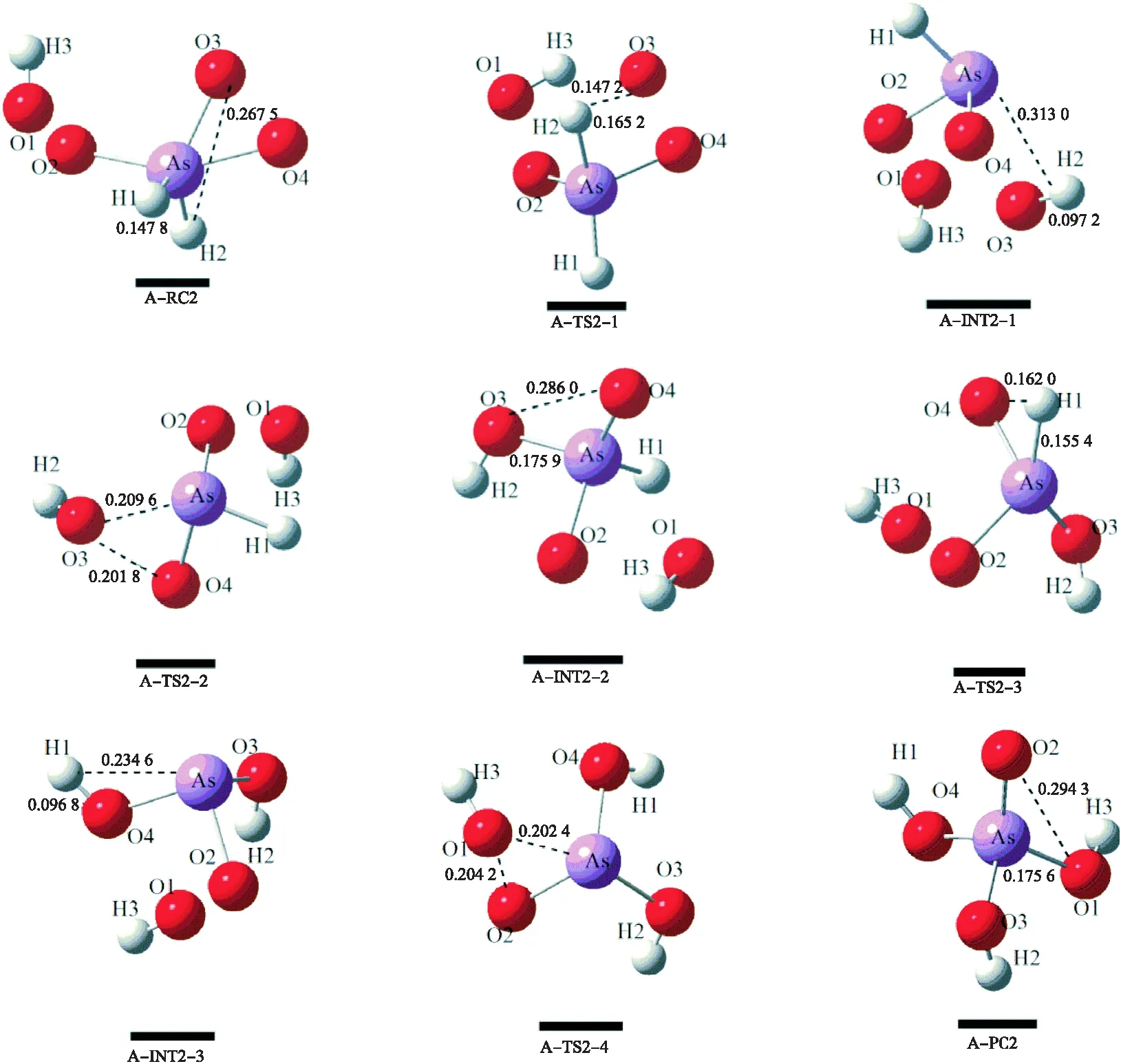
图3 AsH3-2O2直接氧化反应的各驻点在B3LYP/6-31++G(d,p)水平下气相中优化的构型
2.2 途径Ⅲ(AlCl3催化氧化反应)
途径Ⅲ分2步完成.如图4所示,当AsH3中的H1原子接近O1原子的同时,O2原子进攻As原子,得到了过渡态C-TS1,虚频为 -1 191.20 icm-1.在C-RC1,C-TS1及C-PC1中,As-H1的键长分别为 0.147 6、0.160 0 和 0.333 8 nm,O1-H1的键长分别为 0.174 6、0.153 2 和 0.097 5 nm.这表明第1步主要发生As-H1键的断裂和O1-H1键的形成.第2步在反应模型中再加入一分子O2,其过程和上一步的相似,H2原子靠近O3原子,O4原子进攻As原子,得到了C-TS2,对应的唯一虚频是-1 151.65 icm-1.在C-RC2、C-TS2及C-PC2中,As-H2的键长分别是 0.148 6、0.160 8 和 0.333 3 nm,O3-H2的键长分别为 0.260 1、0.154 1 和 0.097 5 nm.该步骤对应着As-H2键的断裂和O3-H2键的形成.
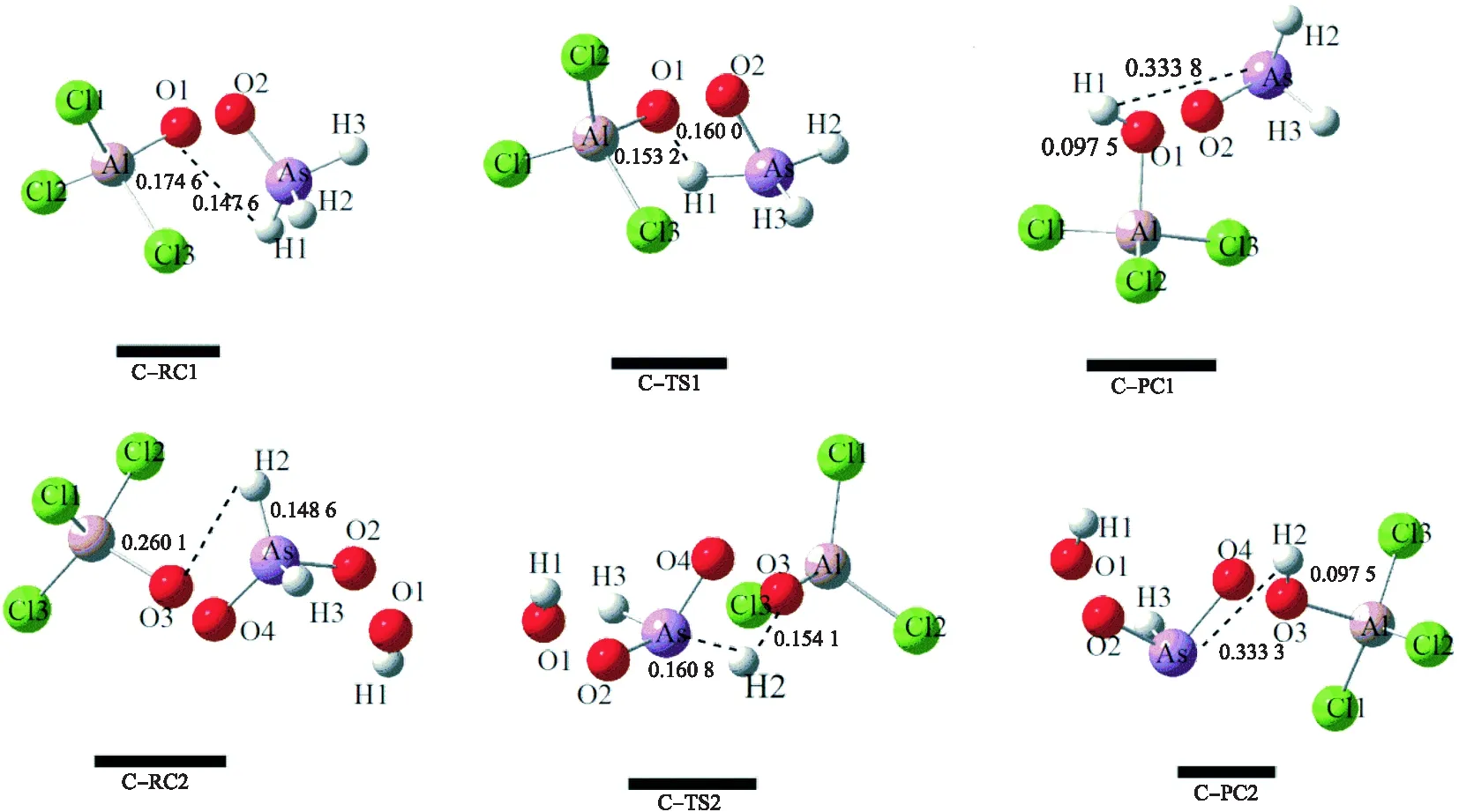
图4 AsH3在AlCl3催化氧化反应的各驻点在B3LYP/6-31++G(d,p)水平下气相中优化的构型
2.3 途径Ⅳ(GaCl3催化氧化反应)
途径Ⅳ反应步骤如下:D-RC1→D-TS1→D-PC1→D-RC2→D-TS2→D-PC2.如图5所示,D-RC1中的As-H1和O1-H1的键长为 0.148 8 和 0.261 1 nm,当H1原子接近O1原子时,O2原子去进攻As原子,得到了过渡态D-TS1,对应的唯一虚频是 -1 164.54 icm-1,其中As-H1和O1-H1的键长为 0.159 3 和 0.154 8 nm,通过振动模式和计算过渡态D-TS1的IRC内禀反应坐标发现从D-TS1→D-PC1的过程中,As-H1从 0.159 3 伸长至 0.338 2 nm;O1-H1从 0.154 8 缩短至 0.097 6 nm.可知第1步反应主要是As-H1键的断裂和O1-H1键的形成.之后再通入一分子O2,得到了D-RC2.在D-RC2、D-TS2和D-PC2中As-H2和分别是 0.148 5、0.160 0 和 0.339 9 nm,O3-H2的键长分别是 0.259 1、0.197 7 和 0.097 7 nm.该步骤对应着As-H2键的断裂和O3-H2键的形成.
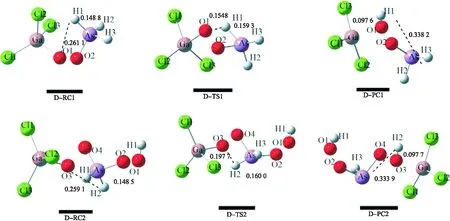
图5 AsH3在GaCl3催化氧化反应的各驻点在B3LYP/6-31++G(d,p)水平下气相中优化的构型
3 能量分析
表1为途径Ⅰ、Ⅱ、Ⅲ、Ⅳ在B3LYP/6-31++G(d, p)水平下得到的相对于分离反应物各驻点能量表(单位:kcal/mol).其中电子能量表示为ΔEe,零点能校正后的能量表示为ΔE0,转动能表示为ΔETRV,焓变表示为ΔH,吉布斯自由能表示为ΔG,自由能垒表示为ΔG≠.
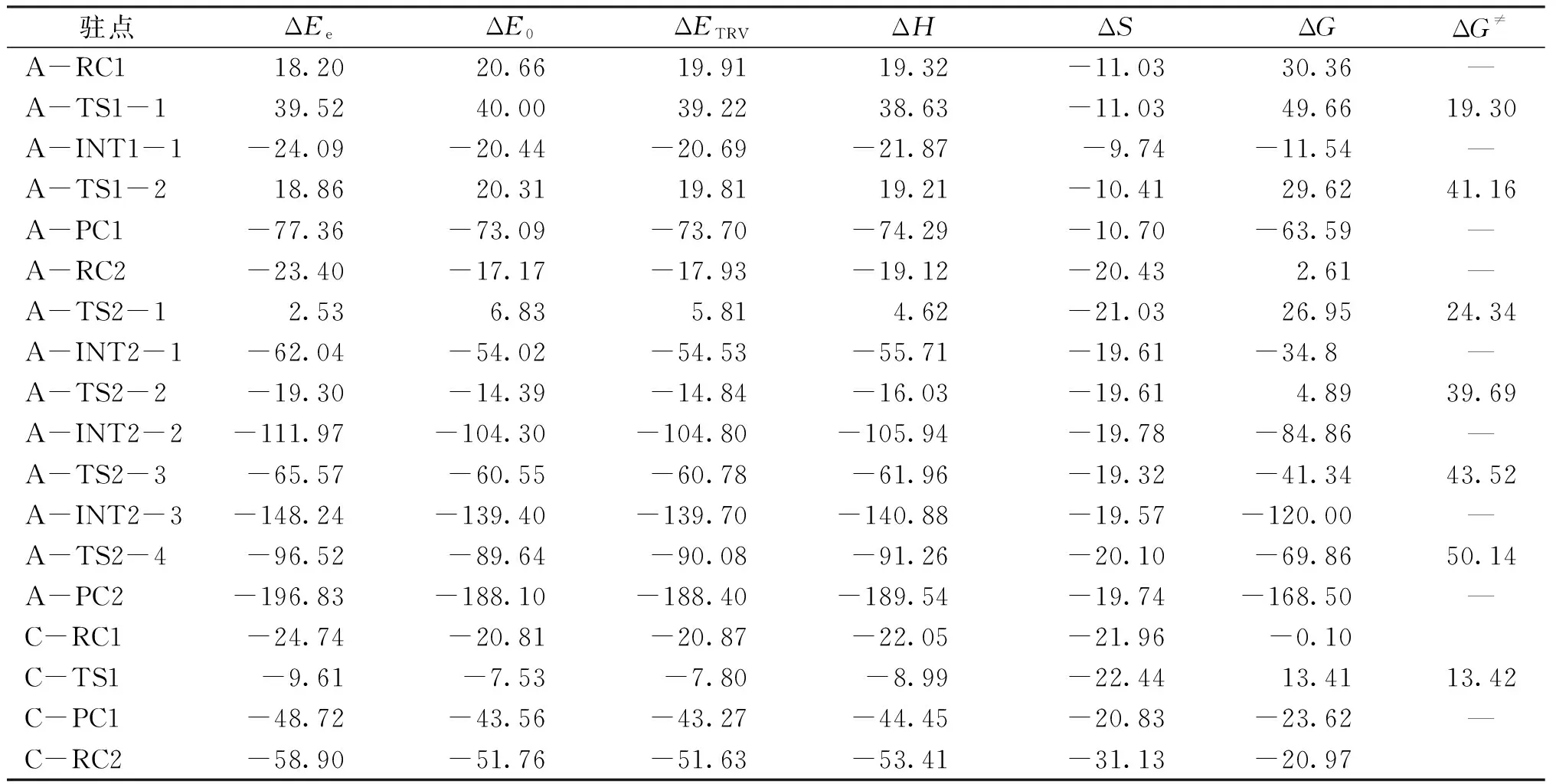
表1 在B3LYP/6-31++G(d,p)水平下得到的途径Ⅰ~Ⅳ的各驻点在气相中分离反应物的相对能量(kcal/mol)
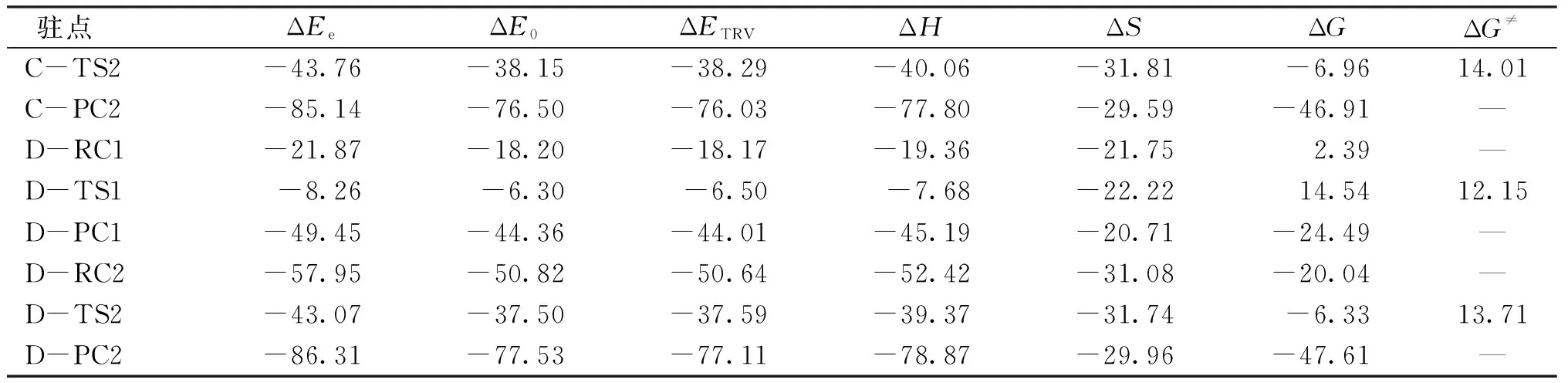
续表1
途径Ⅰ为AsH3与一分子O2反应生成H3AsO2,在该途径中反应物A-RC1的ΔG为 30.36 kcal/mol 、第1个过渡态A-TS1-1的ΔG为49.66.kcal/mol,故第1步的自由能垒ΔG≠为 19.30 kcal/mol.第2步的自由能垒比第1步的自由能垒高,为 41.16 kcal/mol ,所以第2步为速度控制步骤.
途径Ⅱ为H3AsO2与一分子O2反应生成H3AsO4,该途径分4步完成.反应物A-RC2的ΔG为 2.61 kcal/mol、A-TS2-1的ΔG为 26.95 kcal/mol,因此第一步的自由能垒ΔG≠为 24.34 kcal/mol ,后面3个步骤的自由能垒分别为39.69、43.52和 50.14 kcal/mol.由此可见第4步的ΔG≠最高,即该步骤为速度控制步骤.比较途径Ⅰ与途径Ⅱ可知,途径Ⅱ的自由能垒比途径Ⅰ高约 10 kcal/mol,即AsH3与一分子O2反应生成H3AsO2后继续与另一分子O2反应需要克服的能垒更高.
途径Ⅲ为加入催化剂AlCl3后AsH3与O2反应生成H3AsO4,该途径中第1步的自由能垒ΔG≠为 13.42 kcal/mol,与途径Ⅰ第1步(19.30 kcal/mol)相比明显下降.加入了一分子O2后,途径Ⅱ第一步中的ΔG≠为 24.34 kcal/mol,而途径Ⅲ第2步的ΔG≠变为 14.01 kcal/mol,下降了大约 10 kcal/mol.同理,在途径Ⅳ中也有相同的变化趋势,第1步的ΔG≠变为 12.15 kcal/mol,第2步的ΔG≠变为 13.71 kcal/mol.由此可知,催化剂的加入能显著降低AsH3氧化的活化能垒.这可能是因为催化剂中过渡金属的参与让反应体系的电荷分散,而AlCl3和GaCl3分子中心原子有空的p轨道,使O2分子成键轨道向Al或Ga的空p轨道转移,使得O2分子键能削弱,有利于能垒降低.
4 结语
本文采用密度泛函理论研究了砷化氢的直接氧化机理和加入催化剂后催化氧化的反应途径.通过了解反应过程中各驻点的能量、键长以及分子振动状态可知,加入催化剂后,反应的活化自由能垒显著降低.此研究将为AsH3催化氧化反应高效催化剂的寻找和和开发奠定理论基础.
