基于集成学习的N6甲基化位点预测方法研究
2021-04-06赵媛媛陈进祥李富义刘全中
赵媛媛,陈进祥,李富义,吴 昊,刘全中*
(1.西北农林科技大学 信息工程学院,陕西 杨凌 712100;2.蒙纳士大学数据科学中心,澳大利亚 墨尔本 VIC 3800;3.蒙纳士大学生物医学发现研究所和生物化学与分子生物学系,澳大利亚 墨尔本 VIC 3800)
0 引 言
DNA甲基化,是指经DNA甲基转移酶催化,以S-腺苷甲硫氨酸(SAM)作为甲基供体,DNA分子与甲基相连接的过程[1]。在DNA的四种碱基中,只有胞嘧啶和腺嘌呤可以被甲基化。近年来,研究者发现了腺嘌呤的第六位氮原子甲基化修饰,即6-甲基腺嘌呤(N6-methyladenine,6mA)。6mA甲基化作为一种重要的非永久性但相对长期可遗传的基因修饰,被发现在维持细胞正常的转录活性、DNA损伤修复能力、染色质重塑、遗传印记、胚胎发育和肿瘤发生中都有着不可替代的作用,成为分子生物学及医学领域的研究热点[2]。
6mA在DNA层面表达丰富度相对较低,在哺乳动物中,平均每百万个腺嘌呤中只有不到10个6mA位点[3]。目前已经有几种鉴定6mA的实验方法,例如甲基化DNA免疫沉淀测序(MeDIP-seq)[4],毛细管电泳和激光诱导荧光(CE-LIF)[5]和单分子实时测序(MRT-seq)[6]。虽然通过实验方法能鉴定6mA位点,但实验方法实验周期长、劳动强度大且十分昂贵,很难适合从高通量序列中识别6mA。基于机器学习的计算方法可以同时处理多条序列中6mA位点的鉴定,这种方法省时、省力并且效率高,作为实验方法有效的补充,越来越受到生物界的青睐。
最近,华中农业大学周道绣课题组使用了免疫沉淀测序技术对水稻基因组的6mA进行了精确定量和定位,获得了水稻基因组的6mA图谱[7]。该数据的获取为构建基于机器学习模型的6mA识别方法奠定了数据基础。近年来,研究界提出了一些基于传统的机器学习和深度学习的6mA位点预测方法。例如,2019年1月,Chen等提出了一种基于支持向量机方法(6mA-Pred)鉴定水稻因组中的6mA位点[8],模型准确率达到了83.13%。2019年4月,Tahir等人提出了一种卷积神经网络(CNN)计算模型(iDNA6mA)[9],从DNA输入序列中自动地提取关键特征并训练模型,该模型准确率达到了86.59%。2019年7月,Pian基于马尔可夫模型提出了一种新的分类方法(MM-6mAPred)[10],准确率达到89.72%。2019年9月,Liu等人[11]提出了基于提升树模型(ExtraTree)对小鼠和水稻基因中的6mA位点鉴定方法(csDMA),对于水稻中6mA位点的预测达到了86.1%的准确度。
在上述方法中,基于深度学习的iDNA6mA方法不需要人工设计特征,但其识别性能仍有待提高。基于传统机器学习方法的6mA识别方法虽然具有较强的识别能力,但现有的学习模型使用序列要么特征单一,缺乏从多种角度综合考量6mA位点;要么特征维度较高且未使用特征选择方法进行特征选择,如6mA-Pred等,预测的性能还有很大提升空间。
基于上述的现有研究的不足,为了进一步提升6mA位点的预测性能,该研究提出一种基于stacking集成学习的6mA预测模型Stack6mAPred。Stack6mAPred结合了增强核苷酸组成(ENAC)、核苷酸电子-离子相互作用伪电位(EIIP)、核苷酸化学性质(NCP)、Kmer和核苷酸间隔(diTriKGap)5种不同类型的特征编码;利用XGBoost进行特征选择[12],去除冗余特征;集成了朴素贝叶斯、支持向量机(SVM)、LightGBM和逻辑回归等4种不同的分类器。在真实的水稻基因组数据集上进行了实验,结果表明:提出的Stack6mAPred预测模型对6mA位点鉴定的准确率达到91.83%,AUC达到0.967。
1 数据集
数据集构建是机器学习模型的基础,基准数据集的质量对构建模型的性能至关重要。该文使用了Chen等人[8]提供的水稻DNA序列中的6mA数据集。该数据集是从美国国家生物技术信息中心(national center for biotechnology information,NCBI)获得,使用CD-HIT软件[13]去除同源性超过60%的序列。数据集包括880个经实验验证的6mA位点的序列片段和880个非6mA位点的序列片段,序列长度均为41 bp。该数据集已经被多个预测模型使用[8-10]。数据集获取公开站点为http://lin-group.cn/server/ i6mAPred/data。
2 特征提取
不同序列特征对不同问题具有不同的识别能力,最终影响预测模型的性能。为了提取针对6mA位点具有较强预测能力的特征,该研究对iLearn[14]和PyFeat[15]中总结的所有DNA序列特征分别进行性能评估,发现五种对于6mA位点具有较强的识别能力的特征:增强核苷酸组成(enhanced nucleic acid composition,ENAC)、核苷酸电子-离子相互作用伪电位(electron-ion interaction pseudopotentials of trinucleotide,EIIP)、核苷酸化学性质(nucleotide chemical property,NCP)、Kmer、核苷酸间隔(ditriKGap)。特征对应的维度和参数设置如表1所示。
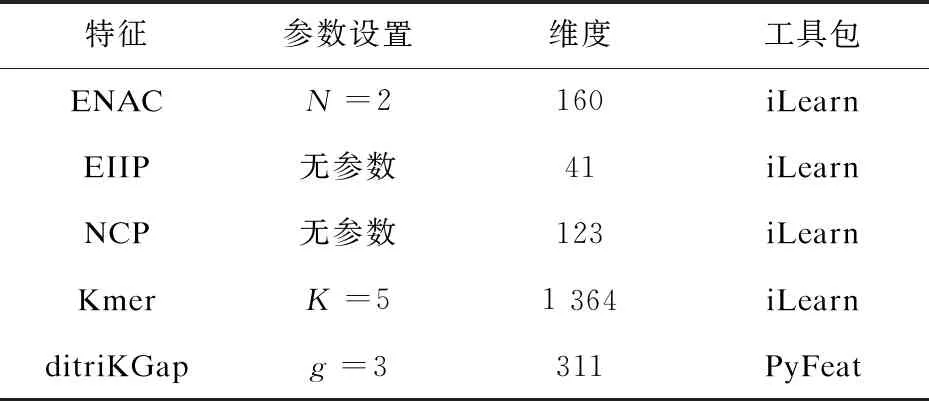
表1 实验中使用的特征及参数介绍
2.1 增强核苷酸组成(ENAC)
增强核苷酸组成(ENAC)根据固定长度的序列窗口计算核苷酸组成(nucleic acid composition,NAC)[16],通常可用于编码等长的核苷酸序列。NAC编码用于计算核苷酸序列中每种核酸类型的频率。序列中四种核苷酸出现频率可以由式(1)计算:
(1)
其中,N(t)是t型核苷酸的数目,N是核苷酸序列的长度。
ENAC编码的核心是计算固定长度的序列窗口内的NAC,即该窗口首先从序列的第一位核苷酸开始,依次向后移动,计算窗口内序列的NAC,直到窗口包含序列的最后一位完成编码过程。实验表明窗口值为2时,ENAC编码的性能达到最优。
2.2 核苷酸电子-离子相互作用伪电位(EIIP)
Nair等人提出了一种新的特征编码方式[17],通过计算核苷酸中离域电子的能量,将其表示为电子-离子相互作用伪电位(EIIP)进行编码。该编码方式直接使用核苷酸的EIIP值取代DNA序列中核苷酸A,G,C和T。核苷酸A,G,C,T的EIIP值分别为0.126 0、0.134 0、0.080 6和0.133 5。EIIP特征编码维数等于DNA序列的长度。
2.3 核苷酸化学性质(NCP)
DNA中有四种核苷酸,即腺嘌呤(A)、鸟嘌呤(G)、胞嘧啶(C)和胸腺嘧啶(T)。根据化学性质进行分类,四种核苷酸分类结果如表2所示。
核苷酸化学性质(NCP)根据每种核苷酸在不同分组内所处类别不同,将每个核苷酸表示为一个3维向量,对特征进行编码。每个化学性质分成两类,一个核苷酸在第1个类中出现编码为1,否则编码为0。因此,根据表2中分类可知,A、C、G和T分别被表示为(1,1,1)、(0,1,0)、(1,0,0)和(0,0,1)。

表2 核苷酸化学性质
2.4 Kmer
Kmer特征编码用于计算DNA序列中K个相邻核苷酸的出现频率,已成功应用于人类基因调控序列预测[18]和增强子识别[19]。Kmer(以K=3为例)由式(2)计算:
(2)
其中,N(t)是Kmer型t的次数,N是核苷酸序列的长度。
文中采用的Kmer编码方式,将小于等于K的相邻核苷酸频率全部计算,以K=3为例,该特征编码将K=3,2,1的Kmer全部列出。经实验验证,K取5使得6mA识别的性能达到最优。
2.5 核苷酸间隔(diTriKGap)
diTriKGap特征编码通过设置间隔大小g,统计DNA或RNA序列内不同间隔序列结构的数目。当设置间隔g=1时,序列结构为XX_XXX;当设置间隔g=2时,序列结构为XX_XXX和XX__XXX;当设置间隔g=3时,序列结构为XX_XXX、XX__XXX和XX___XXX。例如,当设置间隔g=2时,将会统计DNA序列中∑AA_AAA,∑AA__AAA,∑AA_AAC,∑AA__AAC,∑AA_AAG,∑AA__AAG,∑AA_AAT,∑AA__AAT…等结构的数量。特征编码的每一列代表序列结构的数目。
经实验验证,当设置g=3时,diTriKGap特征编码性能达到最优。该编码通过工具包PyFeat提取。为了去除冗余特征,减少特征维度过多[20]而对模型造成的影响,PyFeat采用Adaboost算法进行特征选择,在保持模型性能的同时,将该特征编码的维度由3 072减少到311。
3 特征选择
特征选择是指从初始特征集中选择相关特征子集的机器学习过程,特征选择能有效地降低特征空间的维度,去除对分类不重要的和冗余的特征,提高预测模型的预测性能。
梯度提升决策树(gradient boosting decision tree,GBDT)是一种集成模型,基分类器是CART树[21],适用于分类和回归问题,同时可用于特征选择。XGBoost(extreme gradient boosting)是陈天奇博士在2011年提出的一种基于提升树[21]的集成学习模型。XGBoost是在GBDT的基础上改进,适应范围更广,是对GBDT的一种高效实现。XGBoost中的基分类器是使用CART和线性分类器的组合。
XGBoost根据特征重要性进行排序,以此达到特征选择的目的。如果一个特征在所有决策树中作为划分属性的次数越多,那么该特征就越重要,XGBoost以此计算每个特征的重要性。
在实验中,使用XGBoost进行特征选择,找到最优的特征子集,降低特征维度,流程如图1所示。由于XGBoost模型参数的选择对特征重要性打分影响较大,在实验中改变模型参数进行了36次实验来减少特征选择的误差,最终得到36个不同XGBoost模型的特征打分表,具体步骤如下所述:
(1)将特征编码输入XGBoost模型进行参数打分,得到特征打分表;
(2)重复步骤(1),36次后得到36个互不相同的特征打分表;
(3)将36个特征打分表中的特征取交集,计算交集中每个特征的重要性平均值,并进行排序,得到最终的最优特征子集。

图1 XGBoost特征选择流程
4 基于集成学习6mA位点识别方法
机器学习中的监督学习目标是训练出一个稳定且各方面性能良好的模型。在许多现实场景中,往往只能得到多个有偏好的模型,也就是弱监督模型。根据组合弱监督模型方法的不同,集成学习分为bagging[22]、boosting[23]和stacking[24]三种集成方式。
该研究采用stacking集成学习构建6mA位点预测模型Stack6mAPred,该模型集成4种主流分类器以及5种最优的特征编码构造一个性能更好的6mA位点预测模型。
4.1 模型的整体框架
stacking集成学习模型预测性能的好坏主要取决于基分类器的预测精度和多样性,使用不同参数、不同类型的分类器训练相同的特征,实现不同基学习器之间的强强联合和优势互补。文中提出的stacking集成模型框架Stack6mAPred如图2所示。
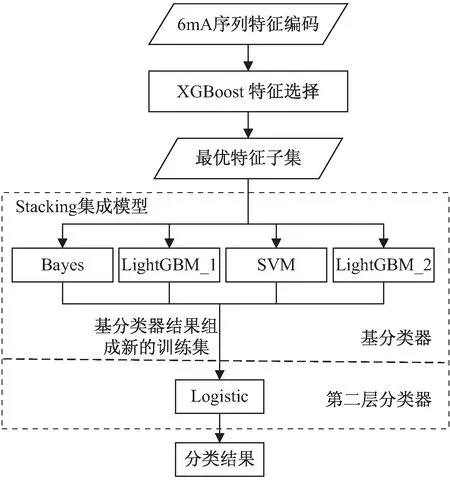
图2 Stacking集成模型框架
初始特征编码由XGBoost进行特征选择得到最优特征子集后,使用stacking集成学习模型训练分类器,得到最终预测结果。本模型中采用4个基分类器,分别由朴素贝叶斯(naive Bayes classifiers,NB)、支持向量机(support vector machine,SVM)和LightGBM等组成,其中LightGBM_1和LightGBM_2均使用LightGBM算法进行训练,两者仅参数设置不同。第二层分类器使用逻辑回归(LR)。
4.2 朴素贝叶斯
朴素贝叶斯分类器假设特征对于给定类的影响独立于其他特征,是一种较稳定的有监督分类算法,其分类算法基于贝叶斯定理,在处理大规模数据库时有较高的分类准确率。贝叶斯分类器的分类原理是通过某对象的先验概率,利用贝叶斯公式计算出其后验概率,即该对象属于某一类的概率,选择具有最大后验概率的类作为该对象所属的类。
4.3 支持向量机
支持向量机是Cortes和Vapnik于1995年首先提出的[25],已经广泛应用于生物信息学问题中。其基本思想是将输入数据转换为高维特征空间,然后确定最佳分隔超平面,以此作为决策边界。
SVM模型有两个非常重要的参数C与gamma。其中C是惩罚系数,即对误差的容忍度,C越高越容易过拟合;gamma是选择RBF作为核函数后,该函数自带的一个参数,决定了数据映射到新的特征空间后的分布。实验中选择RBF作为核函数。
为了选择最优的参数使模型达到最佳性能,使用LibSVM进行参数寻优。LibSVM是用来调整SVM参数非常有效的手段,应用广泛[26-28],该工具包可在https://www.csie.ntu.edu.tw/~cjlin/libsvm/免费获取。
LibSVM采用网格搜索(grid search)进行参数搜索,在C和gamma组成的二维参数矩阵中,依次实验每一对参数的效果,以此得到全局最优的参数。实验最终确定的参数在4.6节进行了详细介绍。
4.4 LightGBM
LightGBM是个快速的、分布式的、高性能的基于决策树算法的梯度提升(gradient boosting)模型[29],是对GBDT的高效实现,可用于排序、分类、回归等机器学习任务中。
该算法在传统GDBT算法基础上引入了梯度单边采样(gradient-based one-side sampling,GOSS)和互斥特征合并(exclusive feature bundling,EFB)两种新技术。梯度单边采样(GOSS)算法保留所有的大梯度样本,在小梯度样本中进行随机采样,从而达到提升效率的目的。独立特征合并(EFB)算法通过使用基于直方图(histograms)方法安全地将互斥特征绑定在一起形成一个新的特征,从而减少特征维度。LightGBM可以在不损失分类器精度的前提下,显著减少模型学习的时间,提升模型的泛化能力。
4.5 集成学习方法
该文采用stacking集成学习模型,其中基分类器使用朴素贝叶斯、LightGBM_1、LightGBM_2和SVM等四个模型,第二层分类器使用逻辑回归。该研究中Stack6mAPred集成模型的训练步骤如下所述:

(3)将D'作为第二层分类器逻辑回归(LR)的训练集,训练得到最终的集成分类模型Stack6mAPred。
4.6 模型参数设置
集成模型中各基分类器中参数的选择对模型分类性能影响极大。在该研究中,为了使集成模型分类性能最佳,先人工确定参数最优的大致范围,然后利用网格搜索(grid search)寻找集成模型的全局最优参数。各基分类器用到的参数如表3所示。

表3 基分类器参数设置
4.7 评价指标
为了评估Stack6mAPred模型的预测性能,分别使用了AUC(area under the ROC curve)值、准确度(accuracy,Acc)、特异性(specificity,Sp)、敏感性(sensitivity,Sn)和马修斯相关系数(Matthews correlation coefficient,MCC)共5个常用的评价指标。其中AUC是ROC(area under curve)曲线与坐标轴围成的面积。ROC曲线是根据一系列不同的二分类方式,以真阳性率为纵坐标,假阳性率为横坐标绘制的曲线。
准确度、特异性、敏感性和马修斯相关系数指标定义如式(3)~式(6)所示:
(3)
(4)
(5)
MCC=
(6)
其中,TP是正确识别的真实6mA序列的数量,FN是错误分类的6mA序列的数量,TN是正确识别的非6mA序列的数量,FP是错误分类的非6mA序列的数量。
5 实验结果
5.1 6mA位点序列分析
为从生物序列方面解释特征编码合理性,本实验使用软件Two Sample Logos[30]绘制6mA位点的序列标识图,该软件对正例样本序列和负例样本序列进行对比,结果如图3所示。

图3 6mA序列对比
从序列标识图可以直观地看出序列共有41个位点,第21号中心点代表6mA位点。每个位点上符号的高度代表对应的核苷酸在该位置的出现频率。
从图3中可以看出,对于6mA位点,在中心位点下游15~18、上游25位置腺嘌呤(A)出现的概率较高,非6mA位点上游22~24位置腺嘌呤(A)出现概率较高,对应文中采用的特征编码ditriKGap中XXX的结构;在中心位点下游20、上游22~23位置鸟嘌呤(G)出现概率较高,非6mA位点下游18~19、14~15等位置鸟嘌呤(G)出现概率较高,对应ditriKGap中XX的结构。由此可以看出,6mA序列具有一定的规律性,反映出文中采用的特征编码中Kmer、核苷酸间隔(ditriKGap)对于提升模型准确度的有效性。
5.2 不同编码方式性能比较
为了找到特征子集的最佳组合,用随机森林分类器(100棵决策树)评估单个特征的性能。表4列举了单个特征以及特征组合预测性能。单个特征中ENAC达到了最佳性能,其次是NCP,而Kmer的性能表现最差。5种特征组合可以显著提高模型的准确度,比单个特征中性能最优的ENAC在敏感性、特异性、准确率、MCC以及AUC值五个性能指标方面分别提高了1.1%、3.2%、2.1%、0.043以及0.02。证明了综合多个特征从多种角度区分6mA位点能提高预测性能。该文整合ENAC、NCP、EIIP、Kmer和diTriKGap 5种特征对样本序列进行编码。
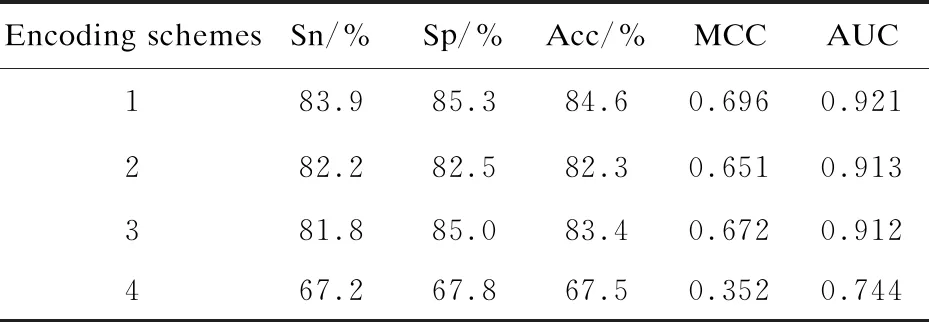
表4 不同编码方式性能比较
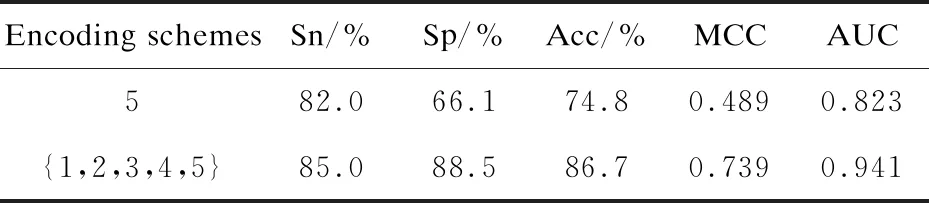
续表4
表4中,编号1、2、3、4和5分别代表五个特征编码ENAC、EIIP、NCP、Kmer和diTriKGap,{1,2,3,4,5}代表五种特征组合。
5.3 特征选择性能评价
为了避免特征冗余以及分类器过拟合,使用了XGBoost进行特征选择,选出最优特征子集。在实验中使用的五种特征编码中,diTriKGap在使用PyFeat进行特征提取时,已经使用AdaBoost进行特征选择。因此,实验仅对ENAC、NCP、EIIP和Kmer的特征组合进行特征选择,将特征选择后的结果与diTriKGap特征编码合并,构成最优特征子集。实验用随机森林分类器(100棵决策树)评估特征选择前后的预测性能。
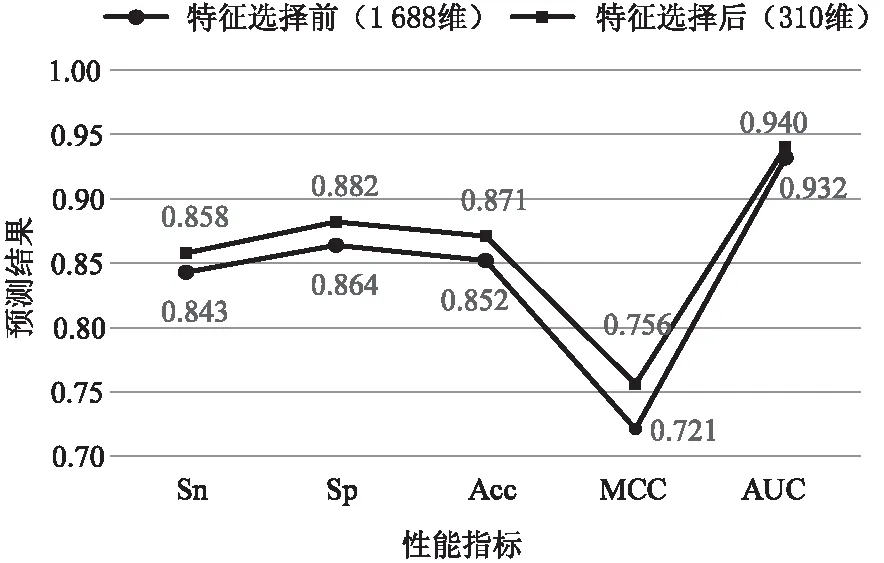
图4 特征选择前后性能对比
实验证明,该文使用的特征选择方法,有效减少了冗余特征,将原来1 688维的特征编码降低为310维。从图4中的五个性能指标来看,该文使用的XGBoost特征选择方法在降低特征编码维度的同时,使模型的性能在所有的评价指标上都有较好的提高。
5.4 不同分类器性能
该文比较了4个不同模型和Stack6mAPred集成模型的分类性能结果。其敏感性、特异性、准确度、MCC、AUC值等5个性能指标对比如图5所示。

图5 不同分类器性能对比
以准确率和AUC值来说,单个模型均达到了较好的性能,准确率都达到了86%以上,AUC值都在0.942以上。其中LightGBM是单个分类器中性能最好的分类器,不同参数的两个LightGBM分类器的准确率分别达到了89.7%和90.4%,AUC值均为0.954;其次支持向量机模型的准确率为90.1%,AUC值为0.946;朴素贝叶斯模型效果最差。与4个单一模型相比,Stack6mAPred集成模型效果最好,最终预测准确率为91.8%,AUC值为0.967,并且敏感性、特异性、马修斯相关系数这些性能指标都有了明显的提升,说明Stack6mAPred集成模型将单一模型优势互补,使预测性能有了较好的提升。
5.5 与现有方法性能比较
为了验证Stack6mAPred模型的性能,将Stack6mAPred与现有模型进行对比。根据调查得知,目前共有4种预测6mA位点的工具:i6mA-Pred[8]、iDNA6mA[9]、MM-6mAPred[10]和csDMA[11]。该研究将Stack6mAPred与以上4种工具从敏感性、特异性、准确度、MCC和AUC这五个指标进行对比分析,比较结果如表5所示。

表5 与现有方法性能比较
从结果中可知,Stack6mAPred预测模型在五个指标上均高于现有工具中最好的MM-6mAPred,其中MCC值提升最为显著,提升了0.06,敏感性、特异性、准确度和AUC分别提升了1.7%、1.36%、1.72%和0.031,这说明了提出的stacking集成方法对于6mA位点预测的有效性。
6 结束语
该文提出了一种基于集成学习的水稻基因组中的6mA位点识别方法Stack6mAPred。该方法组合了5种类型的特征,并且通过XGBoost进行特征选择,去除冗余特征,避免了模型过拟合;集成了朴素贝叶斯、支持向量机、LightGBM和逻辑回归等异构分类器;实现了基分类器之间的强强联合和优势互补,最终构建出一个性能更强的集成预测模型。
该研究以水稻基因组数据为研究对象,构造模型的方法可以迁移到预测其他物种的N6甲基化位点识别中。该研究仅仅集成两层模型,根据需要可以集成更多层的模型,需要指出的是随着模型集成的层次增多,训练的时间会有所增加。
