利用改进的胶束电动毛细管色谱法和液相色谱法测定6类重点食品中的脱氢乙酸
2021-04-03丁晓静林明淑
丁晓静, 林明淑, 张 晶, 苏 萍, 杨 屹
(1. 北京市疾病预防控制中心, 食物中毒溯源技术北京市重点实验室, 北京市预防医学研究中心, 北京 100013; 2. 北京化工大学 理学院, 北京 100029)
脱氢乙酸(DHA)为弱酸,其pKa为5.4[1],难溶于水,易溶于乙腈、甲醇及碱溶液. DHA或其钠盐(DHA-S)[2]对食品中的酵母菌、细菌和霉菌均有抑制作用,我国允许其用于12类食品的防腐[3]. 但过量食用会危害健康. 许多国家对其使用均有严格规定. 美国被允许用于草莓和切或去皮的南瓜,其限量为65 mg/kg. 日本对黄油、奶酪、人造黄油等奶产品的限量为0.5 g/kg, 欧盟则禁止使用[1].
2017年7月,我国宁夏一奶农往牛奶中超范围添加DHA-S,导致多名幼儿发生食物中毒[4-5]. 2019年3月15日,“打假晚会”曝光的、深受学生喜爱的“问题辣条”,严重危害了学生的身体健康[6-7]. 基于此,我国卫生和健康委员会决定重新评估6类重点食品中DHA的安全限量,以保证消费者健康[2]. 而DHA含量的准确测定,对其风险评估具有非常重要的作用.
目前文献报道的DHA的测定方法有气相色谱(GC)法[8-10]、液相色谱(LC)法[8, 11-14]、超高效液相色谱(UPLC)法[15]和毛细管电泳(CE)法[16]. GC法和LC法是我国食品安全国家标准中的测定方法[8],其前处理繁琐费时,LC法[8]中还存在DHA标准溶液配制及流动相不合理的问题,而且均未涉及辣条的测定. CE法[16]同样未涉及辣条和菌类及藻类制品的测定.
本文改进了文献[16]中胶束电动毛细管色谱(MEKC)法和国标中DHA测定的LC法的分离及样品前处理条件,使两种改进后的方法均能准确测定6类重点食品中的DHA. 该法高效、快速、准确且操作简单,非常适合常规实验室大量样品分析.
1 试验部分
1. 1 仪器与试剂
Beckman P/ACE MDQ型毛细管电泳仪具二极管阵列检测器(美国贝克曼库尔特有限公司);50 μm未涂层熔融石英毛细管(河北永年锐沣色谱配件有限公司);Milli-Elix/RiOs超纯水仪(美国Millipore公司);Vortex-Genie 2 涡旋混合器(美国Scientific Industries);KQ-100E医用超声波清洗器(江苏昆山市超声仪器有限公司);Waters 2695高效液相色谱仪具2998二极管阵列检测器(美国Waters公司);Universal 32离心机(德国Hettich);Techmate C18(100 Å,250×4.6 mm,4.6 μm) (北京泰克美科技有限公司).
硼砂(中国医药公司北京采购供应站);硼酸(优级纯,铁岭市开原化工厂);氢氧化钠(优级纯,北京化工厂);十二烷基硫酸钠(SDS,纯度≥99%,Sigma-Aldrich);乙腈和甲醇(色谱纯,Fisher Scientific,USA);脱氢乙酸(纯度≥98%,Sigma-Aldrich);脱氢乙酸钠一水合物(98%,百灵威科技有限公司);磷酸二氢钠、磷酸(国药集团化学试剂有限公司);氢氧化钠(优级纯,北京化工厂);乙腈(色谱纯,Fisher Scientific,USA);山梨酸(≥99%,Sigma-Aldrich);苯甲酸(≥99.5%,Sigma-Aldrich);乙酸铵(分析纯,北京化学试剂公司).
样品:284件,分别从超市、商场、便利店、食品店、菜市场、淘宝网购得,其中咸菜53件,黄油20件,预制肉制品78件,粉丝、粉条及挂面51件,辣条31件,薯片、虾条29件,食用菌及藻类制品22件.
1. 2 MEKC法的10 g/L标准储备液
称取100 mg DHA和113 mg DHA-S,分别置于10 mL容量瓶中,分别加入乙腈、纯水溶解、稀释、定容,涡旋混匀,置于4 ℃冰箱保存.
1. 3 LC法的10 g/L山梨酸、苯甲酸和脱氢乙酸标准储备液
分别准确称取山梨酸和苯甲酸各100 mg,分别使用乙腈溶解、稀释、定容至10 mL,涡旋混匀,置于4 ℃冰箱保存. 10 g/L DHA和DHA-S标准储备液按1. 2节方法制备.
1. 4 样品处理
按文献[17]中调味品的样品前处理方法,即将0.3 g样品置于1.5 mL离心管中,加入0.3 mL饱和NaCl溶液,涡旋混匀后加入0.7 mL乙腈,涡旋混匀,超声提取10 min,9 000 r/min离心5 min,分层后取上层乙腈10 μL,加入190 μL 样品稀释液,混匀后进样. 其中,MEKC法的样品稀释液:1.5 mmol/L 硼砂+ 6 mmol/L硼酸,LC法的样品稀释液:水.
1. 5 改进的MEKC条件
新装毛细管按文献[17-20]方法进行处理.
1. 6 LC条件
改进的色谱条件:色谱柱Techmate C18(100 Å,250×4.6 mm,4.6 μm);流动相为乙腈∶15 mmol/L磷酸+20 mmol/L磷酸二氢钠(体积比为30∶70),等度洗脱;流速:1 mL/min;检测波长:307 nm;柱温:40 ℃;进样量:10 μL.
国标LC条件:色谱柱C18(4.6 mm×150 mm,5 μm);流动相为甲醇∶20 mmol/L乙酸铵(体积比为10∶90),等度洗脱;流速:1 mL/min;检测波长:293 nm;柱温:30 ℃;进样量:10 μL.
2 结果与讨论
2. 1 MEKC法的改进
本实验室前期研究所建立的食品中10种防腐剂的分析方法[16]曾用于能力验证样品测定,获得满意结果,证明了其准确性,故本研究仍用文献[16]所用毛细管规格及分离缓冲体系. 由于本研究测定的食品样品种类及防腐剂种类与文献有所不同,需要在进一步增加分离度及检测灵敏度基础上,着重优化前处理条件.
2. 1. 1 分离缓冲溶液中SDS浓度的优化
文献[16]所用分离缓冲溶液中SDS浓度为100 mmol/L,用于本研究的实际样品分析时,发现存在基质干扰. 保持分离缓冲溶液中60 mmol/L硼酸及15 mmol/L硼砂不变,试验了100、120和140 mmol/L SDS对实际样品中DHA分离的影响,结果如图1所示. 综合考虑分离度及灵敏度,SDS最佳浓度为140 mmol/L. 在此浓度下,本研究所分析的284件实际样品均未发现干扰.

图1 SDS浓度对实际样品中DHA分离的影响 Fig. 1 Effect of SDS concentration on separation of DHA in real samples(a)140 mmol/L, (b) 120 mmol/L, (c) 100 mmol/L Separation buffer: 15 mmol/L sodium borate+60 mmol/Lboric acid+ x mmol/L SDSPeak: (1), (2) unknown, (3) DHACapillary: 50 μm×70 cm (effective length: 60 cm),Separation voltage: 26 kV, injection pressure and time: 3.45×10-3 MPa for 28 s, detection: 228 nm,temperature: 25 ℃
2. 1. 2 进样时间的优化
进样时间对待测物的分离效果和灵敏度有显著影响. 考察了进样压力保持3.45×10-3MPa不变的情况下,进样时间分别为10、20、28和35 s时的分离效果,结果如图2所示. 由图2可见,随着进样时间的增加,各峰校正峰面积明显升高. 样品塞一般占用毛细管有效长度的1%~2%[21],使用CE expert软件计算,进样时间35 s时,样品区带为窗口长度的2.8%,容易导致峰形展宽,分离度减小. 而进样28 s时,样品区带为窗口长度的2.01%. 故最佳进样时间为28 s.

图2 进样时间对DHA分离的影响Fig. 2 Effect of injection time on separation of DHA (a)35 s, (b) 28 s, (c) 20 s, (d) 10 sSeparation buffer: 15 mmol/L sodium borate+60 mmol/L boric acid+ 140 mmol/L SDSOther MEKC conditions and specified peaks were the same as in Fig. 1.
2. 1. 3 样品前处理条件的优化
文献[16]处理咸菜、黄油和调味品等10类食品样品时,分别使用了不同的前处理方法. 故本研究借鉴该文献处理调味品样品的前处理方法,即基于饱和NaCl可使水相与乙腈有机相分层的原理[22],构建相分离体系,拟将6类重点食品样品的前处理方法统一为一种方法. 首先对111#和113#辣条阳性样品进行前处理.
为考察乙腈能否将DHA从样品中一次提取完全,样品均进行第二次提取. 111#样品提取前,样品与离心管总质量1.192 g. 提取完,取出上清液后,样品与离心管总质量为0.799 g,则取出的上清液(乙腈)的质量为:1.192 g-0.799 g=0.393 g. 而加入的0.70 mL乙腈提取液质量为0.544 g,则残留在管内的乙腈质量为0.544 g-0.393 g=0.151 g,残留量为0.151 g/0.544 g=28%. 将第二次提取与第一次提取的校正峰面积相比,比率为52/176=30%. 两个比例相差2%,说明第一次提取时仅有2%未能被提取出,可以忽略,因此认为一次提取便可完全提取样品中的DHA.
同理,113#样品提取前,样品与离心管总质量1.195 g. 提取完,取出上清液后,样品与离心管总质量为0.840 g,则取出的上清液(乙腈)的质量为:1.195 g-0.840 g=0.355 g. 而加入的0.70 mL乙腈提取液质量为0.544 g,则残留在管内的乙腈质量为0.544 g-0.355 g=0.189 g,残留量为0.189 g/0.544 g=35%. 将第二次提取与第1次提取的校正峰面积相比,比率为53/150=35%. 两个比例相同,说明第一次提取即可将DHA提取完全.
市场上可购买到的食品样品中防腐剂均为DHA-S,而非DHA. DHA-S易溶于水而不溶于有机溶剂,为了验证乙腈可以将DHA-S以DHA的形式从食品样品中提取出来,将质量浓度均为10 g/L的DHA标准贮备液和DHA-S标准贮备液稀释100倍后,分别取140 μL加入到称量好的空白样品中,按1. 4节方法处理后进样检测,结果如图3所示. 由图3可见,DHA和DHA-S的定量结果一致,说明这种前处理方法能够将DHA或DHA-S很好的提取出来. 此方法之所以能达到满意的回收率,是因为样品前处理时加入过量饱和NaCl溶液,受盐析效应的影响,DHA-S最终以弱酸形式被提取到乙腈层,从而能够被准确测定[17].
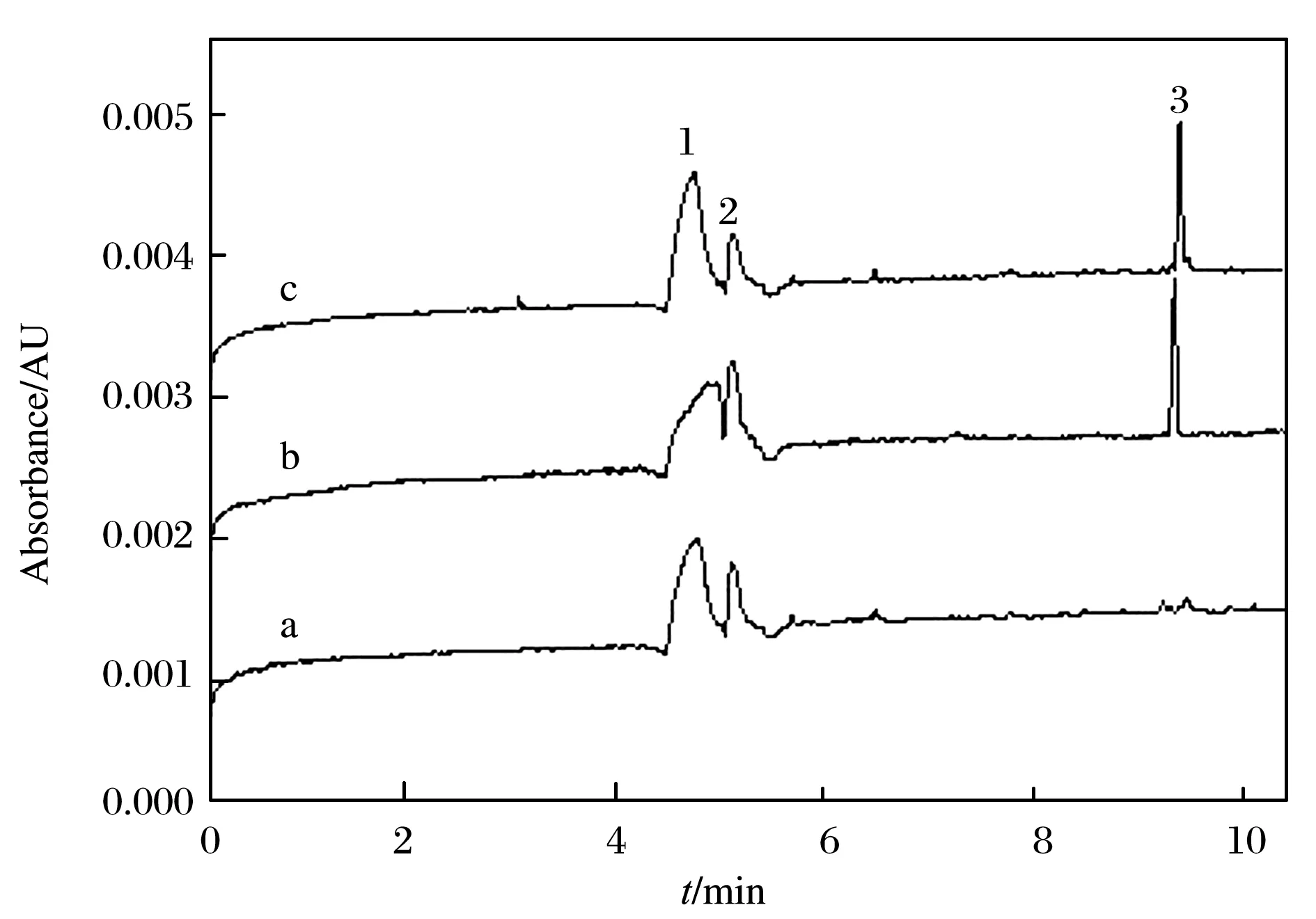
图3 DHA和DHA-S的提取结果Fig. 3 Extraction results of (a) blank sample, (b) sodium dehydroacetate and (c) dehydroacetic acid.Other MEKC conditions and specified peaks were the same as in Fig. 1.
2. 1. 4 标准曲线、线性范围及检出限、定量限
在改进的MEKC条件下,使用乙腈将DHA的储备液(10 mg/mL)逐级稀释,配制混合标准溶液,其质量浓度分别为10、20、40、80、120、200、400、800、1 000 mg/L,然后取每个浓度的混合标准溶液10 μL,再加入190 μL样品稀释液,涡旋混匀,配制成0.5、1、2、4、6、10、20、40、50 mg/L标准系列,保证每个标准溶液中均含5%乙腈. 采用校正峰面积外标法定量,校正峰面积(A)与质量浓度(ρ, mg/L)具有良好线性关系,线性回归方程A=122.219ρ+6.272,线性范围为0.5~50 mg/L,相关系数(r2)为 0.999 9,检出限为0.2 mg/L,定量限为0.5 mg/L.
2. 1. 5 方法精密度
分别平行称取7份咸菜、黄油、预制肉制品、辣条、淀粉制品、藻类及菌类制品样品,分别置于1.5 mL离心管中,按1. 4方法进行处理,在改进的MEKC条件下直接进样测定,计算其含量及迁移时间的相对标准偏差(RSD),方法精密度如表1所列.
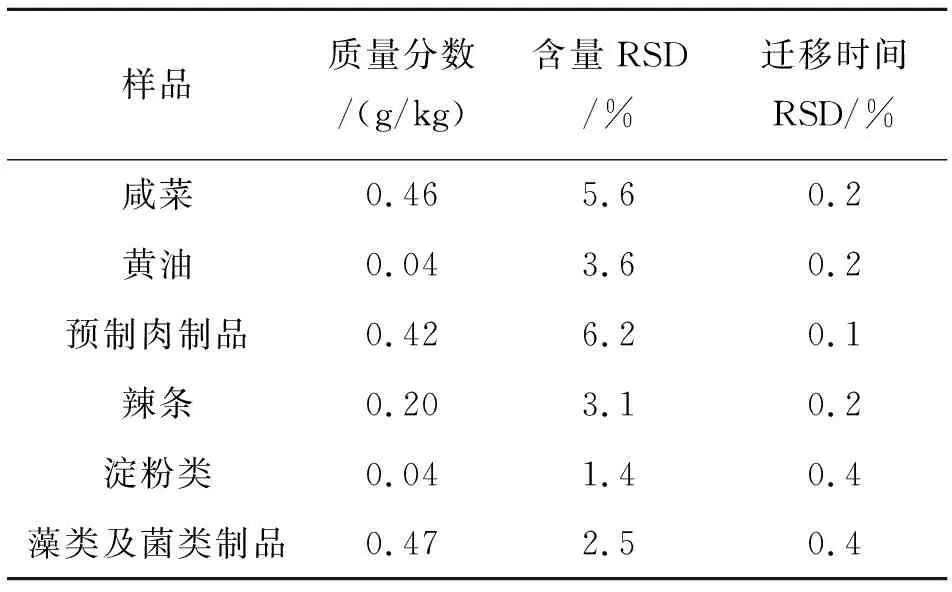
表1 改进的MEKC法测定6类重点食品中DHA方法精密度(n=7)Table 1 Method precisions of DHA in 6 species key food samples by improved MEKC method (n=7)
2. 1. 6 加标回收率
分别选取咸菜、黄油、预制肉制品、辣条、淀粉类制品和藻类及菌类制品的空白样品,分别在1、5、10 mg/L三个水平下进行加标回收率试验,每个加标水平的样品平行处理5份,结果如表2所列. 由表2可见,不同基质样品的低、中、高三个水平的加标回收率在83.7%~116.0%之间,RSD在1.1%~7.3%之间.

表2 改进的MEKC法测定6类重点食品中DHA加标回收率(n=5)Table 2 Results of spiked recoveries of 6 species key food samples by improved MEKC method (n=5) %
2. 1. 7 实际样品测定
使用改进的MEKC法测定购得的284件样品,每件样品均进行两次平行测定. 其中阴性样品207件,小于定量限的有20件. 咸菜中DHA质量分数在0.03~0.40 g/kg之间,辣条在0.05~0.16 g/kg之间,预制肉制品在0.03~0.40 g/kg之间,藻类及菌类制品在0.02~0.34 g/kg之间,淀粉类仅有一件检出,质量分数为0.48 g/kg. 阳性样品均未超标.
2. 2 LC法的改进
2. 2. 1 储备液介质的选择
DHA的lgKow经EPI Sutie 4.1软件计算为0.78,25 ℃时水中溶解度为690 mg/L[23],微溶于水. 故国标法中的LC法配制DHA标准储备液时,使用氢氧化钠溶解,根据配制方法可知:DHA标准系列质量浓度最低点1 mg/L中氢氧化钠浓度为0.05 mmol/L,理论计算标准溶液pH≈9.7,最高质量浓度点中氢氧化钠浓度为10 mmol/L,理论计算此标准溶液pH≈12,如此高pH易导致硅胶填料的色谱柱柱效下降. 故本研究采用乙腈溶解DHA并制备储备液.
2. 2. 2 流动相的优化
食品中常用防腐剂除DHA外,还同时添加山梨酸或苯甲酸,因LC的分离柱效较MEKC低2~3个数量级[24],为避免这两种防腐剂的干扰,将3种防腐剂配制成混合溶液,优化流动相条件. DHA pKa为5.4[1]、山梨酸和苯甲酸的pKa均为4.2[16]. 理论上,流动相的pH应不高于pKa-2,即酸性流动相体系才能保证三者以分子形式存在而且能够在反相C18色谱柱上保留,从而实现与杂质分离,得到较为准确的定量结果. 当采用国标中偏弱碱性的流动相甲醇∶20 mmol/L 乙酸铵(体积比10∶90)时,峰形拖尾严重,如图4所示,且分析实际样品时DHA与干扰峰的分离不达基线,影响其准确定量. 当采用文献[25]所用流动相0.02 mol/L乙酸铵(氨水调pH为9.0)∶甲醇(体积比97∶3)时,DHA峰形略有前伸,如图5所示. 当采用0.015 mol/L磷酸+0.02 mol/L磷酸二氢钠∶乙腈(体积比70∶30)时,DHA峰形较好,且时间较短,如图6所示. 故选择0.015 mol/L磷酸+0.02 mol/L磷酸二氢钠∶乙腈(体积比70∶30)为最佳流动相.

图4 三混标在国标法的流动相条件的色谱图Fig. 4 Chromatogram of three mixed standards in mobile phase of national standard Peak: (1) benzoic acid, (2)sorbic acid, (3) DHA Column:techmate C18 (100 Å, 250×4.6 mm, 4.6 μm),mobile phase: V(methanol)∶V(20 mmol/L ammonium acetate)=10∶90, flow rate: 1 mL/min, detection: 293 nm
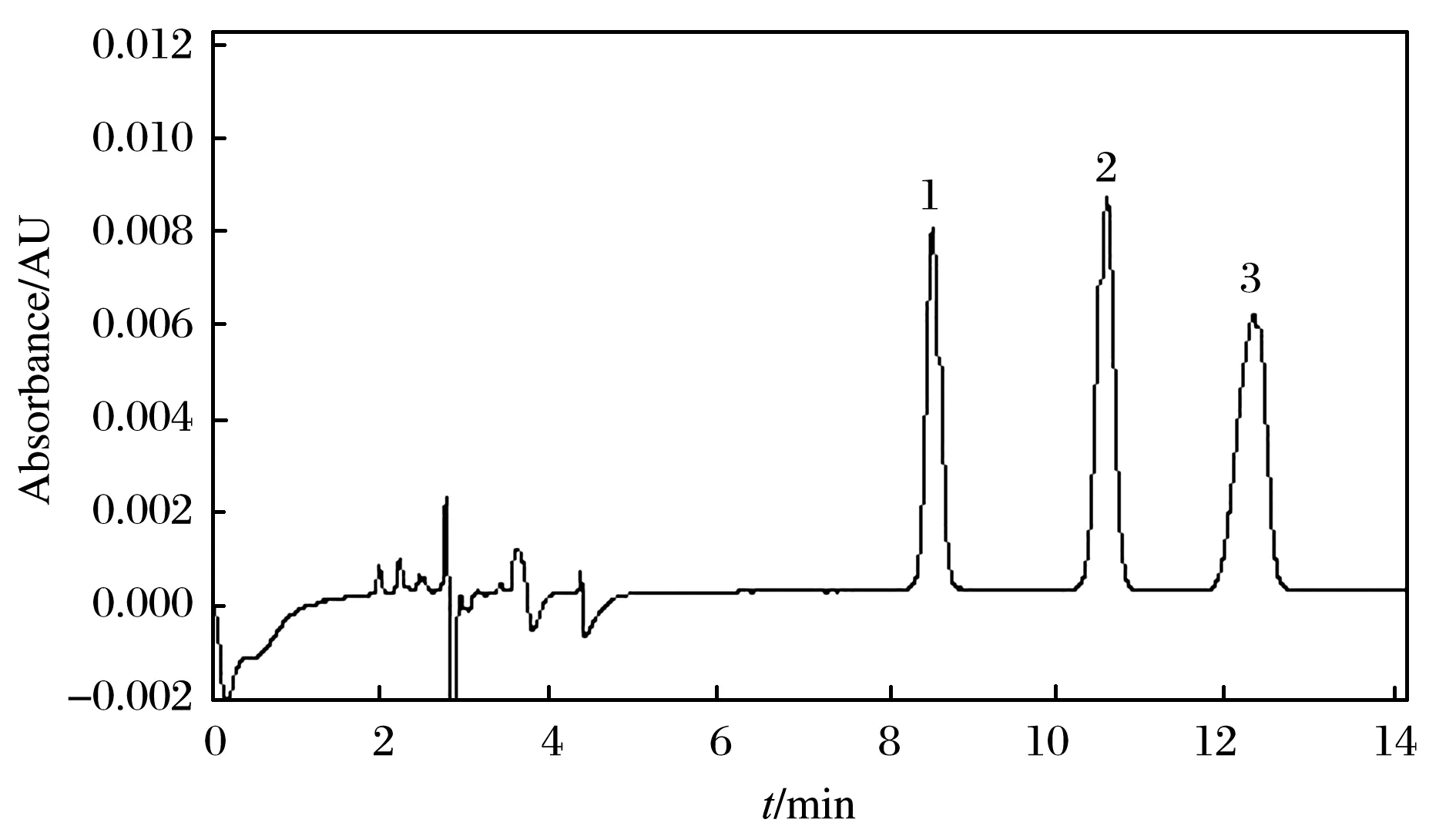
图5 三混标在文献[25]所用流动相的色谱图Fig. 5 Chromatogram of three mixed standards in mobile phase of reference[25]Mobile phase: 0.02 mol/L ammonium acetate, pH 9.0 adjusted with ammonium hydroxideOther LC conditions and specified peaks were the same as in Fig. 4.

图6 三混标在含磷酸盐缓冲体系流动相的色谱图Fig. 6 Chromatogram of three mixed standards in mobile phase of phosphate buffer solutionMobile phase:V(acetonitrile):V(15 mmol/L phosphate acid+20 mmol/L sodium dihydrogen phosphate, pH 2.34)=30∶70, detection: 307 nmOther LC conditions and specified peaks were the same as in Fig. 4.
DHA的最大紫外吸收波长与流动相的pH有很大关系. 当流动相为弱碱性的乙酸铵体系时,分别在230、291 nm有最大吸收,如图7所示. 在流动相pH为2.34的15 mmol/L磷酸+20 mmol/L磷酸二氢钠体系中,分别在223、307 nm有最大吸收,但307 nm的紫外吸收峰高于223 nm,如图8所示,故本研究选择307 nm为检测波长.

图7 DHA在乙酸铵流动相中紫外吸收光谱图Fig. 7 Ultraviolet absorption spectrum of DHA in mobile phase of ammonium acetateOther LC conditions and specified peaks were the same as in Fig. 4.
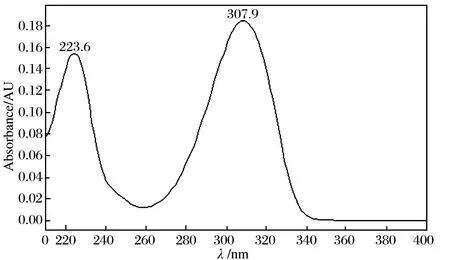
图8 磷酸盐流动相中DHA紫外吸收光谱图Fig. 8 Ultraviolet absorption spectrum of DHA in mobile phase of phosphate as shown in Fig. 6Other LC conditions and specified peaks were the same as in Fig. 4.
2. 2. 3 样品稀释液的优化
与MEKC法的样品前处理一样,乙腈提取后,需要使用样品稀释液对乙腈提取液进行稀释. 分别考察水、3%乙腈-水、5%乙腈-水、10%乙腈-水溶液以及5%乙腈-15 mmol/L 磷酸+20 mmol/L 磷酸二氢钠这5种稀释液对DHA分离及峰高的影响,结果表明:这5种样品稀释液对DHA的分离及峰高无明显影响,故以水为样品稀释液.
2. 2. 4 前处理方法的改进
国标法[8]中DHA的LC法的样品前处理通过调节样品溶液pH,将DHA-S提取出来,易损失和沾污样品. 此外,不同类别的样品需采用不同的前处理方法,大部分样品需固相萃取柱净化,操作复杂繁琐费时,根据误差传递理论,前处理方法步骤越多,则结果的误差也越大. 此外消耗的试剂(特别是有机溶剂)、耗材(容量瓶、离心管等)较多,而且中间还有调节pH的环节,容易造成样液的损失而导致结果不准确. 与MEKC法一样,充分利用DHA易溶于乙腈的性质,构建了利于乙腈提取的条件,将加入的DHA-S盐,通过盐析作用,最终以DHA的形式被乙腈提取出来. 为进一步确认盐析作用,取两份0.5 mL 2 mg/L DHA-S(水介质),分别加入0.5 mL乙腈. 其中一份溶液中加入过量NaCl固体至不溶,则溶液分层,如图9(a)所示. 另一份溶液中不加NaCl固体,则溶液不分层,如图9(b)所示. 加入饱和NaCl固体的溶液中DHA的峰面积大约是不加NaCl固体的两倍,说明盐析作用效果明显.

图9 NaCl对DHA提取的影响Fig. 9 Effect of NaCl on extraction of DHA(a) 0.5 mL acetonitrile was first added to 0.5 mL 2 mg/L DHA-S, and then NaCl was added until saturated NaCl solution was formed, (b) 0.5 mL acetonitrile was added to 0.5 mL 2 mg/L sodium dehydroacetate without NaCl addition Peak: (1) DHAOther LC conditions were the same as in Fig. 6.
2. 2. 5 标准曲线、线性范围及检出限、定量限
在改进的LC条件下,使用乙腈将DHA的储备液(10 g/L)逐级稀释,其质量浓度分别为4、10、20、40、80、120、200、400、800、1 000 mg/L,然后每个浓度取10 μL,再加入190 μL 水,配制成0.2、0.5、1、2、4、6、10、20、40、50 mg/L标准系列. 采用峰面积外标法定量,峰面积(A)与质量浓度(ρ, mg/L)具有良好线性关系. 线性回归方程A=2.61×104ρ+38.1,线性范围为0.2~50 mg/L,相关系数(r2)为0.999 9,检出限为0.05 mg/L,定量限为0.2 mg/L.
2. 2. 6 方法精密度
分别平行称量7份咸菜、黄油、预制肉制品、辣条、淀粉制品、藻类及菌类制品样品,分别置于1.5 mL塑料离心管中,按1. 4节方法处理,使用改进的LC法测定,方法精密度结果如表3所列.

表3 改进的LC法测定6类重点食品中DHA方法精密度(n=7)Table 3 Precisions of DHA in 6 species key foods by improved LC method (n=7)
2. 2. 7 加标回收率
选取咸菜、黄油、预制肉制品、辣条、淀粉类、藻类及菌类制品样品的空白基质,分别在低、中、高3个质量浓度水平(1、5、10 mg/L)进行加标回收率试验,每个水平的样品平行处理5份,结果如表4所列.

表4 改进的LC法测定6类重点食品中DHA加标回收率(n=5)Table 4 Results of spiked recoveries of 6 species key food samples by improved LC method (n=5) %
2. 2. 8 实际样品测定
为进一步验证改进后的LC法的可靠性,分别使用国标液相色谱法和改进的液相色谱法,分别测定国标前处理方法和改进的前处理方法处理的样品. 鉴于国标前处理法中调节pH时,需将电极直接插入样品溶液,易污染电极,最终缩短电极使用寿命,造成较高的检测成本,在此只将一件咸菜样品用于方法比对. 按国标色谱条件,分别按国标及改进的前处理方法得到的色谱图如图10所示. 由图10可见,使用国标液相色谱条件测定时,无论是国标前处理还是改进的前处理方法,都有杂质未被完全分离,并且定量结果的相对标准偏差为7.2%. 当用改进的液相色谱条件时,无论是国标前处理还是改进的前处理方法,分离均好,如图11所示,且定量结果的相对标准偏差减小,为2.9%,二者结果基本吻合,再次证明国标方法在分析样品基质相对简单的咸菜样品时,无论是色谱分离还是样品前处理,均存在不合理之处,需改进.
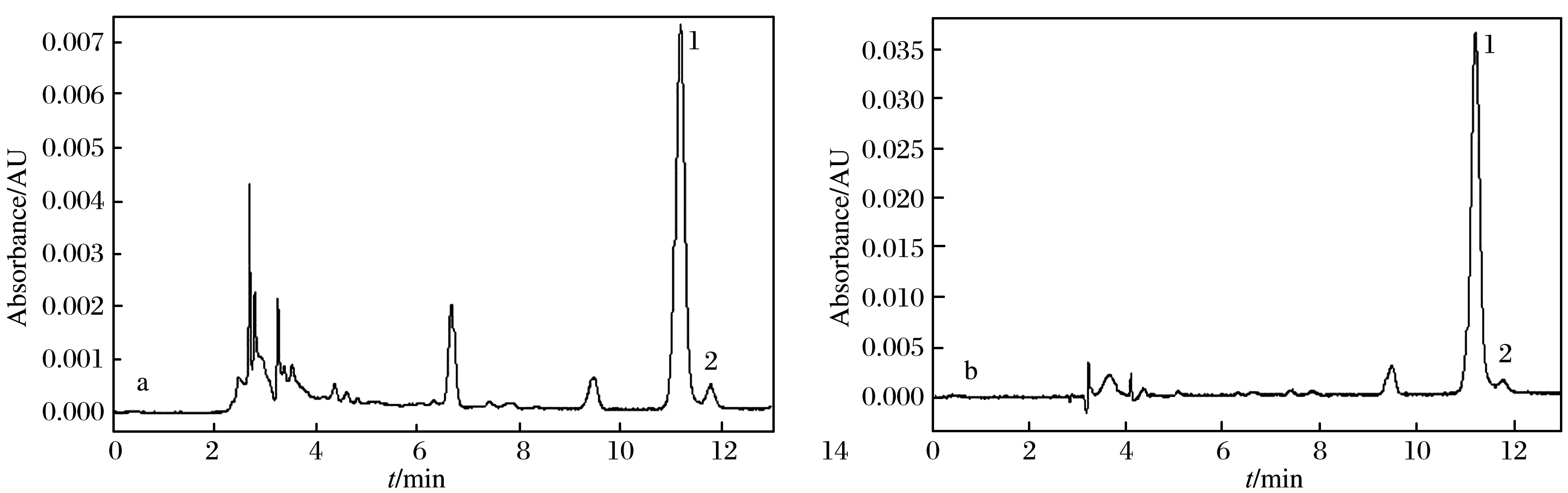
图10 国标液相色谱条件下,分别按国标和改进的前处理方法处理咸菜样品的色谱图Fig. 10 Chromatograms of pickle samples treated according to national standard and improved pretreatment method, respectively under chromatographic conditions of national standard (a) sample pretreatment method according to national standard method, (b) improved sample pretreatment methodPeak: (1) DHA, (2) unknownOther LC conditions were the same as in Fig. 4.
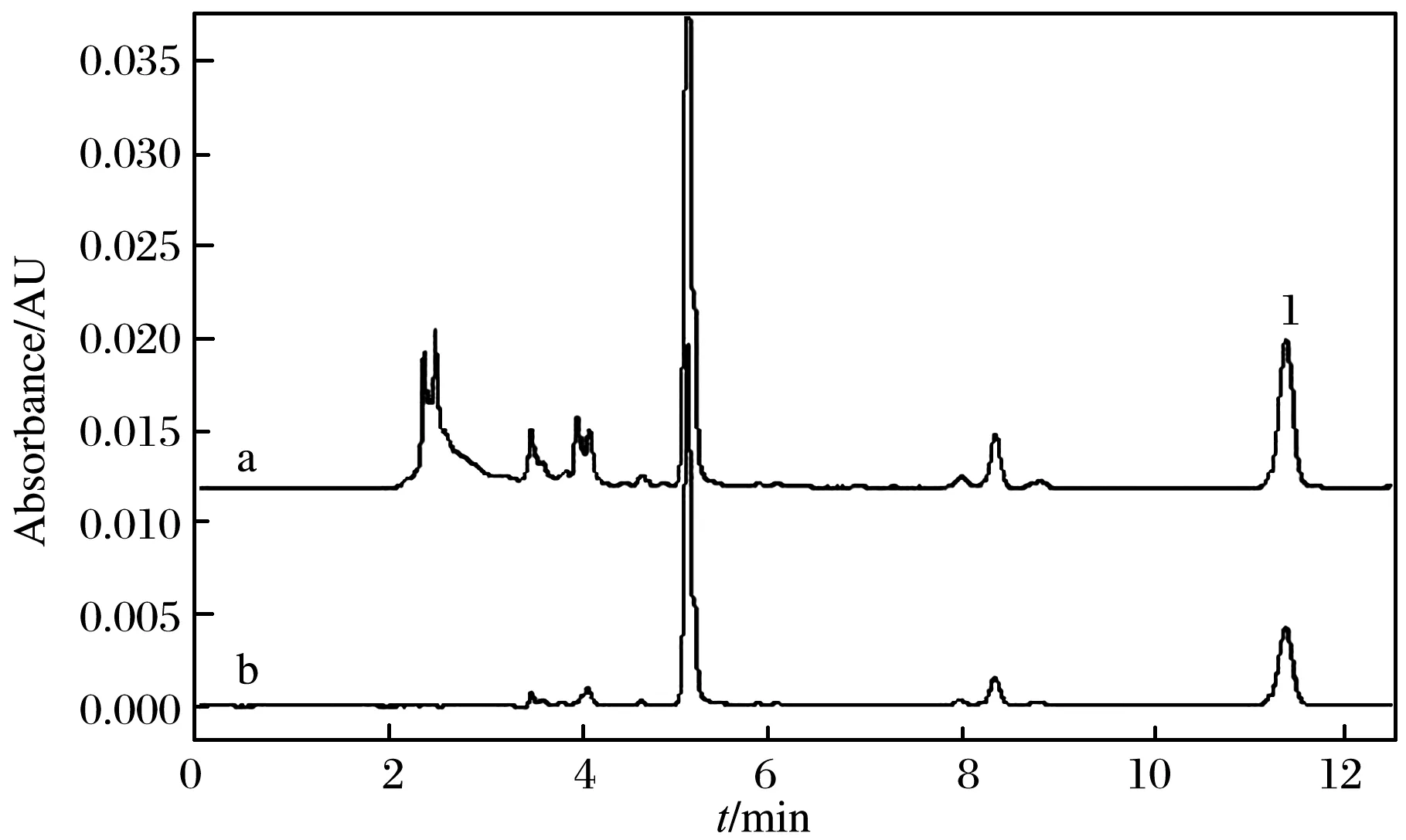
图11 改进的国标液相色谱条件下,分别按国标和改进的前处理方法处理咸菜样品的色谱图Fig. 11 Chromatograms of pickle samples treated according to national standard and improved pretreatment method, respectively under chromatographic conditions of improved national standard (a) sample pretreatment method according to national standard method, (b) improved sample pretreatment method in this researchPeak: (1) DHAOther LC conditions were the same as in Fig. 6.
使用改进的国标方法对45件样品进行了检测,并与改进的MEKC结果比对,其中25件样品两种方法结果均为阴性,另外20件阳性样品结果如表5所列. 可见,两种改进方法的测定结果吻合好,适用于6类重点食品样品的测定.

表5 改进的MEKC和改进的LC法对阳性样品的测定结果Table 5 Results of positive samples by improved MEKC and LC methods /(g/kg)
3 结论
分离是定量的基础,分离好,定量结果才能准. 但如果实际样品的前处理不当,干扰物质不能被去除,则在分离时将产生干扰,导致定量不准确. 改进后的两种方法,无论是从分离还是样品前处理,均比原来的方法更科学合理,这已在SDS和流动相优化的条件试验中得以体现. 因为改进后的前处理方法通过盐析效应,仅将溶于乙腈的组分从样品基质中有效提取,284件实际样品的测定结果表明,干扰被有效消除,故能够得到准确的分析结果.
将6类重点食品的前处理方法统一为一种方法. 将改进的MEKC法与改进的国标LC法测定咸菜样品的结果进行了比较,二者吻合性好,进一步验证了两种改进方法的准确性. 将改进的MEKC法用于6类重点食品的检测,加标回收率均在83.7%~116.0%之间,相对标准偏差在1.1%~7.3%之间. 无论是改进的MEKC还是LC法,前处理均简单、易行. MEKC法的检测成本低,更适合大量样品的分析.
