小分子脾酪氨酸激酶抑制剂临床研究进展
2021-04-01范露崔兵兵陆涛陈亚东
范露,崔兵兵,陆涛,陈亚东
(中国药科大学理学院,江苏 南京 211198)
1 脾酪氨酸激酶概述
B细胞抗原受体(B-cell antigen receptor,BCR)信号传导在细胞增殖、分化、迁移和存活中起重要作用,其参与信号级联反应主要是由3个非受体型酪氨酸激酶家族介导:Zeta链相关的蛋白激酶(zeta-chain-associated protein kinase,ZAP)、Src家族(proto-oncogene tyrosine-protein kinase SRC,原癌基因酪氨酸蛋白激酶SRC)和酪氨酸蛋白激酶家族(tyrosine-protein kinase,Tec)。其中ZAP家族包括脾酪氨酸激酶(spleen tyrosine kinase,Syk)与Zeta链相关的蛋白-70(zeta-chain-associated protein kinase-70,ZAP-70);Src家族包括:重组人酪氨酸蛋白激酶Lyn与Fyn;Tec家族包括:布鲁顿酪氨酸蛋白激酶(bruton's tyrosine kinase,BTK)、白细胞酪氨酸激酶(leukocyte tyrosine kinase,LTK)和内皮酪氨酸激酶(endothelial tyrosine kinase,ETK)。作为BCR信号转导通路中的重要信号分子,Syk是蛋白酪氨酸激酶家族(protein tyrosine kinase,PTK)中的一员,其与ZAP-70同属一个PTK家族并且在结构上高度同源。Syk广泛表达于肥大细胞、嗜碱性粒细胞、B细胞和血小板等造血细胞中,其编码基因为Syk,位于人类染色体9q22[1],相对分子质量为72 000,由629个氨基酸组成。该酶由2个Src同源2结构域(N-SH2和C-NH2)、一个C端激酶结构域和域间A/B组成。域间A位于2个SH2 结构域之间,域间B位于SH2结构域和C端结构域之间[2](见图1)。在静息状态下,Syk保持未被磷酸化的状态。当BCR、Fc受体(Fc receptor,FcR)、T细胞抗原受体(T-cell antigen receptor,TCR)等免疫受体受到抗原、病原体相关分子模式(pathogen-associated molecular pattern,PAMP)、损伤相关的分子模式(damage-associated molecular pattern,DAMP)等因素刺激后,首先Lyn磷酸化免疫受体酪氨酸激活 基 序(immunoreceptor tyrosine-based activatory motif,ITAM)上的2个酪氨酸残基。双磷酸化的 ITAM与Syk的SH2结构域结合,从而在胞内招募Syk[3]。随后Syk磷酸化激酶结构域上的酪氨酸残基Y525/Y526[4](人序列)和域间A上的调控残基Y130,导致Syk从自抑制状态转变为活化状态[5];同时Syk暴露停泊位点,进而与含SH2结构域的白细胞蛋白-65(SH2 domain-containing leukocyte protein-65,SLP-65,又称BLNK)结合使得活化构象得以稳定。
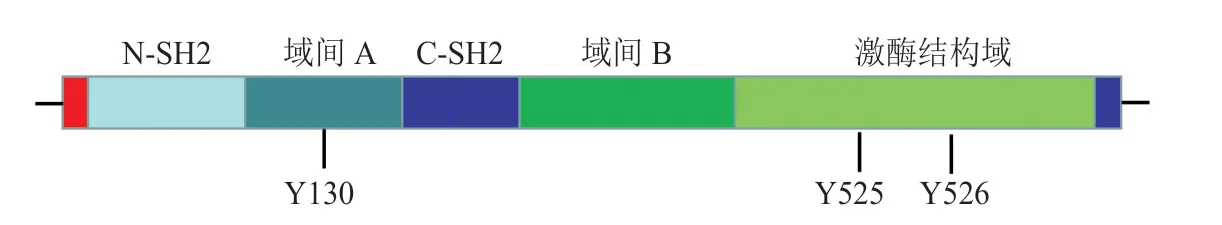
图1 Syk的结构Figure 1 The structure of Syk
Syk被激活后,进一步磷酸化多个信号配体,激活下游一系列信号通路,例如控制细胞增殖的大鼠肉瘤病毒同源癌基因激酶(rat sarcoma viral oncogenehomolog,RAS)-快速加速的纤维肉瘤激酶(rapidly accelerated fibrosarcoma,RAF)-有丝分裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)-激 活 蛋 白1(activator protein 1,AP1)信号通路;控制细胞存活以及细胞因子释放的BTK-磷脂酶Cγ2(phospholipase Cγ2,PLCγ2)-三 磷 酸 肌 醇(inositol triphosphate,IP3)-活化T细胞的Ca2+-核因子(Ca2+-nuclear factor of activated T cells,NFAT) 信 号 通 路或BTK-PLCγ2-蛋 白 激 酶C(protein kinase C,PKC)-核 转 录 因 子-κB(nuclear factor κB,NFκB)信号通路;调节细胞生长的磷脂酰肌醇-3-激酶(phosphatidylinositol 3-kinase,PI3K)-蛋白激酶B(protein kinase B,AKT)-哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)信号通路(见图2)。这些信号通路在细胞生长、增殖和细胞因子释放等方面具有重要作用。
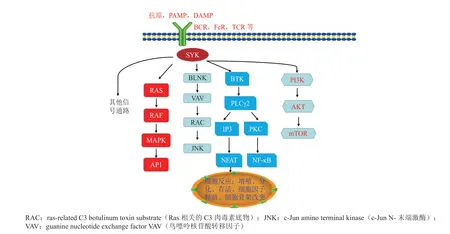
图2 Syk影响的信号通路Figure 2 Syk-affected signaling pathways
2 脾酪氨酸激酶与疾病治疗
Syk位于信号通路上游,调节下游众多信号通路,在促进细胞因子和化学因子等炎症因子分泌以及B细胞的增殖、分化、存活过程中扮演关键的角色,被认为是肿瘤和自身免疫等疾病治疗的重要靶标。
2.1 抗肿瘤治疗
BCR信号异常激活是B细胞恶性肿瘤的主要病理机制,B细胞恶性肿瘤包括慢性淋巴细胞性白血病(chronic lymphocytic leukemia,CLL)、弥漫性大B细胞淋巴瘤(diffuse large B-cell lymphoma,DLBCL)、急性髓细胞白血病(acute myeloid leukemia,AML)和套细胞淋巴瘤(mantle cell lymphoma,MCL)等。BCR通过Syk的组成型信号 传 导 激 活BTK- NF-κB和PI3K-AKT-mTOR信号通路[6],促进B细胞增殖、存活等。鼠类研究表明Syk表达是非霍奇金淋巴瘤(non-Hodgkin lymphomas,NHL)体外存活所必需的,在体外药理抑制Syk活性会诱导小鼠B细胞淋巴瘤细胞凋亡,从而导致NHL样B细胞淋巴瘤消退[7]。在一部分DLBCL细胞系中,基础水平和BCR诱导的SLP-65磷酸化均可诊断细胞对R406(1)抑制Syk的敏感性,并与R406对这些细胞的抗增殖作用相关。同样,以Syk的磷酸化增强为特征的BCR信号与临床上更具侵袭性的CLL相关,在没有抗体刺激的情况下,CLL细胞中的Syk及其下游信号通路激酶磷酸化水平较高。用R406处理原代CLL细胞可减少Syk磷酸化以及下游信号转导事件,例如AKT和细胞外调节蛋白激酶(extracellular regulated protein kinases,ERK)的磷酸化以及骨髓细胞白血病蛋白-1(myeloid cell leukemia-1,Mcl-1)抗凋亡蛋白的表达。在这些原代CLL细胞中,Syk的抑制导致细胞凋亡,这突显了这些抑制剂在CLL治疗中的潜在用途[8]。临床试验结果也证明Syk抑制剂无论是单独使用或与其他治疗方式联用均能阻断BCR信号通路从而抑制肿瘤的增殖和生长[9-10]。在实体瘤方面,最新的研究显示Syk和表皮生长因子受体(epidermal growth factor receptor,EGFR)的过度活化有关,在鳞状细胞癌的发展中起着重要的作用[11]。因此Syk是治疗多种肿瘤的潜在靶点,其治疗应用有待进一步开发。
2.2 自身免疫性疾病的治疗
目前常见的自身免疫性疾病包括类风湿性关节炎(rheumatoid arthritis,RA)、系统性红斑狼疮(systemic lupus erythematosus,SLE)、1型糖尿病(type 1 diabetes,T1D)、特发性血小板减少紫癜(idiopathic thrombocytopenic purpura,ITP)等。在自身免疫性疾病中,适应性免疫的成分错误地将自身抗原识别为外源抗原,并启动了消除此类抗原程序。ITP和自身免疫性溶血性贫血(autoimmune hemolytic anemia,AIHA)特点在于血小板和红细胞被破坏,其是通过自身抗体识别细胞特异组分,导致黏膜皮肤出血和贫血。而RA和SLE是由于自身抗体识别因体积太大而无法被巨噬细胞和中性粒细胞吞噬的自身抗原引起的。在这种情况下,吞噬细胞将诸如活性氧(reactive oxygen species,ROS)、蛋白酶和磷脂酶之类的有毒物质释放到环境中,引起细胞和组织的局部破坏,目的是在一些附带损害的情况下中和“有害”物质。最终产生并释放脂质介质以发出疼痛信号。趋化因子和细胞因子的产生将更多的免疫细胞募集到损伤部位,建立起协调一致的持续性炎症反应,从而加剧了组织的炎症反应,最后导致永久性结构损伤。
自身免疫性疾病的发生和免疫球蛋白G-Fc受体γ(immunoglobulin G-FcRγ,IgG-FcRγ)信号通路的异常激活息息相关。IgG-抗原复合物结合免疫细胞上的FcRγ(FcγRⅠA、FcγRⅡA、FcγRⅢA)受体从而介导炎性免疫反应。FcRγ受体通过ITAM或半免疫受体酪氨酸激活基序(hemi-immunoreceptor tyrosine-based activation motif,hem-ITAM)介导激活Syk,进而增强吞噬细胞对抗原的吞噬作用,刺激细胞产生细胞因子、NO和ROS,促进杀死微生物并引起组织损伤[12]。由于Syk在IgG-FcRγ信号通路中的关键作用,其被认为是治疗自身免疫性疾病的有效靶点。有研究显示缺乏Syk的巨噬细胞不能吞噬IgG包被的颗粒,而缺乏Syk的中性粒细胞无法响应FcγR的信号而发生氧化性破裂,因此Syk抑制剂可以阻断上述IgG-FcRγ信号过程的发生而治疗自身免疫性疾病。
2.3 过敏性疾病的治疗
在发达国家高达25%的人口患有过敏性疾病,包括过敏性鼻炎、哮喘和特异性皮炎(atopic dermatitis,AD)等。在分子水平上,这些疾病主要是由过敏原特异性免疫球蛋白IgE和TH2辅助细胞驱动。致敏受试者单次暴露于特定过敏原会导致早期反应或Ⅰ型即时超敏反应,这是过敏原通过IgE交联FcεRⅠ受体导致肥大细胞和嗜碱性粒细胞发生脱粒引起的。肥大细胞和嗜碱性粒细胞脱粒导致分泌组胺、类胰蛋白酶、5-羟色胺和肝素,随后数分钟后产生并释放类花生酸、脂质介质(白三烯和前列腺素),然后数小时后产生趋化因子和炎性细胞因子。这些介体共同刺激白细胞的募集和激活,以进行后期变态反应,包括TH2细胞、嗜酸性粒细胞、嗜碱性粒细胞和其他白细胞的募集和激活。不断暴露于特定的过敏原会通过先天和适应性免疫细胞导致慢性过敏性炎症,从而导致组织重塑。目前治疗哮喘、过敏性鼻炎和特应性皮炎的方法包括限制接触有害的过敏原以及使用抗组胺药、局部色酮和皮质类固醇。尽管在现有疗法中取得了成功,但某些患者的症状仍然持续,因此有足够的机会开发进一步的新颖疗法。肥大细胞的过度活化和脱粒导致多种促炎性介质的释放是过敏性疾病的关键组成部分,因此这些过程的抑制剂代表了开发新疗法的潜在途径。在此过程中,IgE交联FcεRⅠ受体导致其多聚化并快速引发ITAM磷酸化,双磷酸化的ITAM募集并激活Syk。Syk进一步磷酸化PLCγ、BLNK,激活下游信号通路,导致细胞因子分泌或NFAT、NF-κB、AP1转录因子激活从而引发机体的炎症反应[13]。因此过敏反应中大部分信号介体的释放受到Syk控制,抑制Syk不仅可以阻止肥大细胞和嗜碱性粒细胞的活化,而且可以阻止巨噬细胞、中性粒细胞、嗜酸性粒细胞的抗原和免疫球蛋白定向活化。此外除了阻止最初的肥大细胞对过敏原的有害反应,Syk的抑制还是影响多种后续抗原特异性炎症反应的关键节点。临床上越来越多证据显示通过靶向Syk抑制IgE介导的过敏反应效果显著[14],Syk已经成为治疗急性和慢性过敏性炎症的有效靶标之一。
3 Syk抑制剂的临床研究进展
鉴于B细胞受体信号在多种疾病中的重要的作用,新型Syk 抑制剂研究已日益引起人们的注意,小分子Syk抑制剂在近几年得到了快速的发展,随着进入临床的药物不断增多,多个小分子Syk抑制剂已展现出开发潜力。本文综述了目前已上市或在全球处于Ⅱ期临床阶段的Syk抑制剂的研发进展(见表1)。

表1 Syk抑制剂研发进展Table 1 Development progress of Syk inhibitors
3.1 Fostamatinib
Fostamatinib(R788,2)是由里格尔制药(Rigel Pharmaceuticals)研发的非选择性口服Syk抑制剂,对Syk的解离常数Kd为15 nmol · L-1,对另外25个激酶的Kd小于15 nmol · L-1以及对54个激酶的Kd小于100 nmol · L-1[15],包括Janus激酶2(Janus kinase 2,JAK2)、FMS样酪氨酸激酶3(Fms-like tyrosine kinase,FLT3)和血管内皮细胞生长因子受体2(vascular endothelial growth factor receptor 2,VEGFR2)等激酶。Fostamatinib作为前药在体内可通过肠碱性磷酸酶转化为活性疏水性代谢物R406,该代谢物迅速吸收进入血液循环发挥作用[16],其最大血药浓度(Cmax)为550 ng · mL-1,药物浓度-时间曲线下面积(AUC)为7 080 ng · h · mL-1,达到(Cmax)的时间(Tmax)为1.5 h,药物半衰期(t1/2)为15 h[17]。Fostamatinib最初开发的适应证为RA,但是由于其脱靶效应以及剂量毒性等问题,针对RA的临床试验都被暂停或终止。目前fostamatinib已于2018年在美国批准上市,用于治疗对既往治疗反应不足的慢性和难治性ITP[18]。这是目前唯一被批准上市的Syk抑制剂。在2项Ⅲ期临床多中心、随机、双盲、安慰剂对照试验(FIT1和FIT2)中,具有持续性/慢性ITP的患者按2∶1的比例随机分配给药fostamatinib(n= 101)和安慰剂(n= 49),注射24周(100 mg,bid),无反应者的剂量于4周后增加150 mg(bid)。主要终点指标是稳定的反应(在14 ~ 24周不进行急救治疗的情况下,病人血小板计数在6次双周访视中有超过4次大于50 000 µL-1)。接受fostamatinib治疗的患者中有18%出现了稳定的应答,而安慰剂组则为2%。fostamatinib治疗组患者总体反应率占43%,而安慰剂组患者占14%。中位反应时间为15 d(100 mg, bid),83%的患者在8周内有应答。最常见的不良事件为腹泻、高血压、恶心和头晕等。大多数不良事件为轻度或中度,可自发解决或通过药物治疗解决。Fostamatinib对ITP患者产生了具有临床意义的反应,包括脾切除术、血小板生成药物和奥利妥昔单抗治疗失败的患者[19-20]。
除了治疗ITP,fostamatinib目前还被开发更多的适应证。3项关于自身免疫性疾病的临床试验正在进行中,分别为第3阶段持续性/慢性ITP患者扩展试验,自身免疫性溶血性贫血患者的SOARⅡ期临床试验以及IgA肾病患者的SIGNⅡ期临床试验[17,21]。除此之外,fostamatinib可以选择性地抑制BCR信号通路从而抑制B细胞肿瘤生长。在CLL和DLBCL的小鼠模型中,其可以诱导正常和恶性B细胞的早期和短暂动员,但选择性抑制恶性B细胞群的生长。这种对B细胞动员的影响类似于BTK抑制剂[22],因此fostamatinib在血液肿瘤中的临床应用有待于进一步研究。

3.2 Entospletinib
Entospletinib(GS-9973,3)是由吉利德科学公司开发的一款高选择性Syk抑制剂。它对Syk的Kd为7.6 nmol · L-1,除了对端锚聚合酶(tankyrases,TNK)的Kd小于100 nmol · L-1,对其他激酶的Kd均大于100 nmol · L-1。相比于R406,人们希望高选择性的entospletinib可以带来更好的治疗前景。CLL细胞系的临床前研究表明,抑制Syk活性可导致趋化因子配体3(CCL3)和CCL4分泌减少,从而导致细胞重新分布。同时破坏了B细胞和周围基质的微环境,减轻了趋化因子和整联蛋白介导的CLL细胞的保护作用[23-24]。临床上,这种作用表现为淋巴结减少以及相关的短暂性淋巴细胞增多。这些作用使entospletinib成为复发或难治性CLL(R/R CLL)和其他NHL的理想药物,所有被治疗患者都显示肿瘤体积减小[25]。接受entospletinib一项多中心Ⅱ期临床研究评估了患有R/R CLL(n= 41)和NHL(n= 145)的受试者(800 mg,bid)治疗的有效性和安全性。。24周时的无进展生存期(PFS)为70.1%,中位PFS为13.8个月;客观缓解率(objective response rate,ORR)为61%,其中3例(ORR 为7.3%)通过持续性淋巴细胞增多获得淋巴结缓解。54名受试者(ORR 为29%)出现严重不良事件(SAE)。最常见的治疗性SAE包括呼吸困难、肺炎、发热性中性粒细胞减少、脱水和发热。由此看出entospletinib在具有可接受毒性的R/R CLL受试者中证明了其临床活性[15]。此外研究证明在FLT3-ITD(内部串联重复突变)的AML细胞系中,entospletinib可以阻止Syk的组成型磷酸化和MYC蛋白表达[26],其对AML的治疗具有重要意义。关于AML的Ⅰb/Ⅱ期临床研究结果显示无论单药治疗还是联合化疗都表现出良好的耐受性。3名患者接受200 mg(bid),6名接受400 mg(bid)治疗。最常见的非血液学SAE包括发热性中性粒细胞减少、恶心和腹泻。综合所有数据,选择400 mg(bid)作为Ⅱ期临床试验推荐剂量。出乎意料的是在2个水平上治疗的患者(9/9,100%)均达到完全缓解(complete response,CR)。一名11q23基因重排的AML患者在接受entospletinib单药治疗14 d后达到形态学和细胞遗传学CR,表明该患者对这种药物具有独特的敏感性[1,27]。
Entospletinib对R/R CLL和AML的临床表现显示出Syk抑制剂对部分B细胞肿瘤的治疗潜力,但其在对惰性非霍奇金淋巴瘤(iNHL)和MCL的Ⅱ期临床评估中表现出相对低的反应率,缺乏足够有效性[28]。目前临床试验正在评估entospletinib与长春新碱和奥比妥珠单抗联用在更大范围的B细胞恶性肿瘤中的作用。
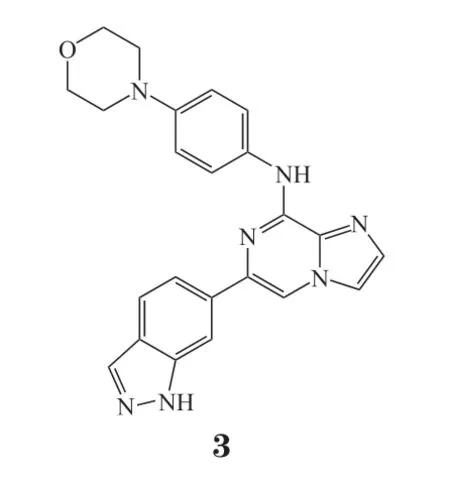
3.3 Lanraplenib
Lanraplenib(GS-9876,4)是在entospletinib的基础上开发的针对自身免疫性疾病的Syk抑制剂。Entospletinib虽然在血液肿瘤的临床评估中取得了较好的疗效,但是该药的pH依赖性溶解度影响其吸收并导致和质子泵抑制剂(PPI)的药物相互作用以及其bid给药方案这些因素限制了它在治疗炎症等疾病方面的应用。Lanraplenib在结构改造过程中利用氨基吡嗪替代了吲唑环而减少了芳香环的数目,降低了平面性从而实现了溶解度的提升。同时使用N-氧杂环丁烷哌嗪替代容易被氧化代谢的吗啉环,从而提升了人的肝微粒体稳定性。此外在激酶竞争性结合实验中表现出低于10%的结合率(只对395个激酶中的12个显示出较好结合能力)。Kd测定结果显示lanraplenib对11个脱靶激酶中的8个Kd均在Syk的10倍以上。由此可见lanraplenib保持了entospletinib的高选择性。在大鼠狼疮模型中,用lanraplenib进行处理可预防与疾病进展相关的蛋白尿增加,并改善动物的存活率,它的功效与使用环磷酰胺达到的功效相当。Lanraplenib在健康人体内的安全性和药代动力学特征评估中,志愿者接受2 ~ 50 mg的单次剂量以及15 ~ 50 mg的多次剂量后耐受良好,无SAE或临床报告的明显实验室异常。由此看出lanraplenib具有良好的药代动力学特性,适合qd给药,并且与PPI没有任何相互作用[29]。
Ⅱ期临床评估了lanraplenib对有甲氨蝶呤(MTX)治疗背景的活动期RA患者的安全性和有效性,结果显示和安慰剂相比并没有显著的临床有效性[30]。由于lanraplenib的单药治疗缺乏足够有效性,临床正在评估lanraplenib和JAK抑制剂filgotinib联用对中度至重度活跃性皮肤红斑狼疮(CLE),狼疮性膜性肾病(LMN)和活动性干燥综合征等自身免疫性疾病的安全性和有效性。
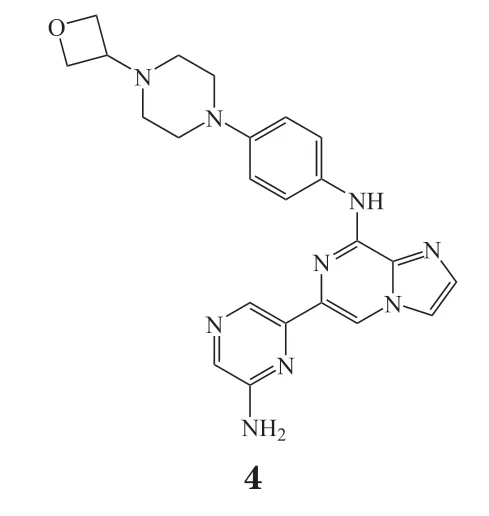
3.4 Cerdulatinib
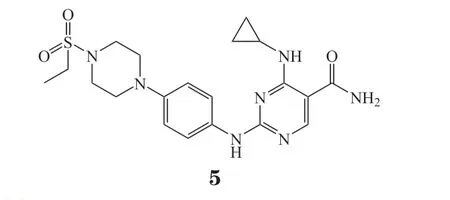
Cerdulatinib(PRT062070,5)是 由Portola Pharmaceuticals公司研发的Syk/JAK双靶点抑制剂,其对Syk、JAK1/2/3和TYK2(tyrosine-protein kinase 2,酪氨酸激酶2)的IC50分别为32、12、6、8和0.5 nmol · L-1[31]。单独靶向B细胞信号通路的BTK抑制剂ibrutinib,在治疗包括CLL和MCL在内的B细胞肿瘤时显示出较好的疗效,但其在临床可达到的浓度下不会显著诱导CLL细胞死亡,因此根除残留疾病的可能性很低,甚至出现耐药性等问题。而cerdulatinib相比ibrutinib能够克服微环境的支持并诱导CLL细胞死亡,并且阻断了对ibrutinib耐药的原代CLL细胞和经BTKC481S转染的对ibrutinib耐药的淋巴瘤细胞的增殖[32],因此可能会克服其耐药问题。另外临床前研究结果表明cerdulatinib相对于单独的Syk或JAK抑制剂都显示出更高的抗肿瘤活性[33-34]。
在自身免疫性疾病和B细胞肿瘤动物模型中证明了cerdulatinib的有效性。经口给药后,cerdulatinib在胶原诱导的大鼠关节炎模型中抑制了炎症和自身抗体的产生,并在慢性B细胞抗原受体刺激的小鼠模型中阻断了B细胞的活化和脾肿大。在DLBCL的模型评估中,cerdulatinib诱导了ABC和GCB淋巴瘤细胞系的凋亡和细胞周期停滞,阻断了GCB和非GCB DLBCL肿瘤细胞中的JAK/信号传导及转录激活因子(signal transducers and activators of transcription,STAT)和BCR信号传导[1,35]。Ⅰ期临床试验评估了cerdulatinib对复发和难治性B细胞肿瘤的有效性和安全性。2名滤泡性淋巴瘤(FL)患者最初在前2个治疗周期内接受本品45 mg(bid)给药方案后达到了CR,5名患者(CLL,n= 3;FL,n= 1;转化3B级FL,n= 1)获得了部分缓解(partial response,PR)[36]。总体来说,cerdulatinib耐受性良好,并且在接受过大量先期治疗的复发/难治性B细胞恶性肿瘤患者中显示出较为理想的抗肿瘤活性。Cerdulatinib临床进展最快的适应证为周围T细胞淋巴瘤(PTCL),在2018年美国FDA已授予它治疗PTCL的孤儿药地位,目前正处于Ⅱ/Ⅲ期临床评估中。除了PTCL之外,正在进行的Ⅱ/Ⅲ期临床评估还包括CLL、小淋巴细胞淋巴瘤(SLL)和NHL等患者中的剂量递增研究[36]以及0.37%的cerdulatinib凝胶对白癜风成年患者治疗的安全性和有效性研究。
3.5 ASN002
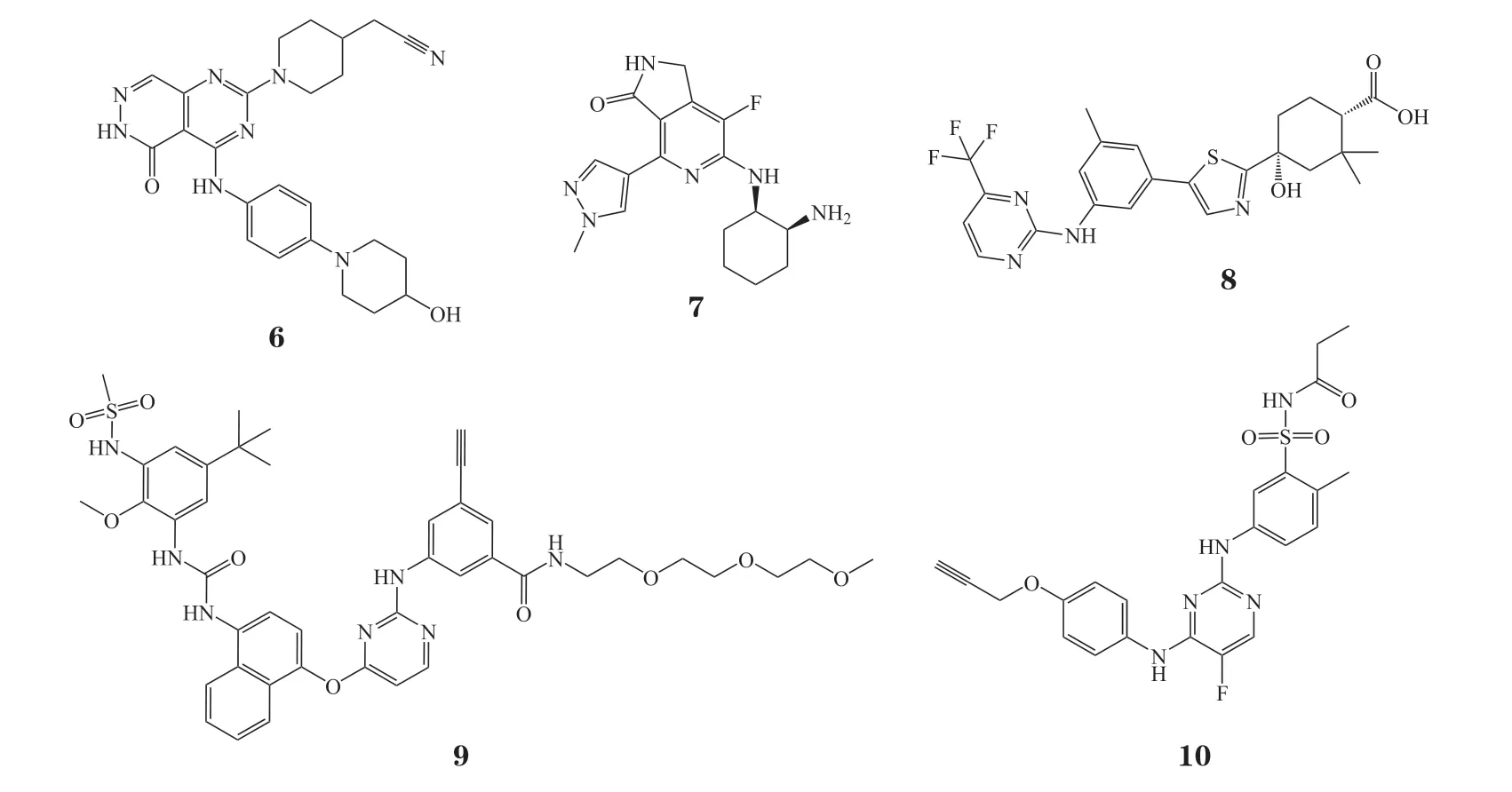
ASN002(6)是Asana BioSciences公司研发的口服JAK/Syk双重抑制剂,其对Syk、JAK1/2/3和TYK2的IC50分别为5、46、4、11和8 nmol · L-1。它的适应证是AD。AD是全球范围内广泛流行的慢性疾病,整体来看,AD在成年人中的发生率达5% ~ 10%,儿童中更达到10% ~ 15%[37]。然而目前针对AD的系统治疗措施依旧有限,当前以润肤剂、类固醇、钙调神经磷酸酶抑制剂和免疫抑制剂的主流治疗方法的疗效有限,且可能产生严重的不良反应[38],并且非常缺乏口服药物。ASN002可以降低各种在皮肤炎症中产生作用的蛋白的活性,是首个被证实可改善AD皮损表型相关临床症状的口服药物。
一项针对AD的Ⅰb期临床研究中评估了ASN002的有效性和安全性。36名中度至重度AD患者被随机平均分为安慰剂组和不同剂量(20、40、80 mg,qd)组,持续4周治疗。结果显示在湿疹面积和严重程度指数(EASI)评估中,40、80 mg 剂量组和安慰剂组实现EASI改善50%的患者比例分别100%、83%和22%,实现EASI改善75%的患者比例分别71%、33%和22%。ASN002耐受性良好,在所有组别中,SAE一般较轻且相似,并且更高剂量的 ASN002未使不良反应的频率或严重程度增加。此外ASN002还显示出剂量依赖性血浆暴露,且患者间变异性较低,有效抑制AD有关炎症通路和临床反应,显著下调了涉及TH1、TH2和TH17/22 等致病有关的几种血清生物标志物水平[39],同时降低了与动脉粥样硬化相关的生物标记物E选择素(SELE)表达[40]。ASN002 治疗还可快速改善瘙痒程度,导致与炎症相关的血液循环蛋白的水平显著降低。除了针对AD治疗,目前Ⅱ期临床还在评估ASN002与Hedgehog通路抑制剂(vismodegib)联用在基底细胞癌治疗中的有效性和安全性。
3.6 TAK-659
TAK-659(7)是由武田制药公司研发的Syk/FLT3双重抑制剂,其对Syk和FLT3的IC50分别为3.2和4.6 nmol · L-1,对JAK3、VEGFR2和ZAP-70具有20倍以上的选择性[41]。TAK-659可以抑制微环境引起的促生存、促增殖、化学耐药性[1]。TAK-659与其他BCR抑制剂的组合在诱导凋亡方面显示出协同作用,它和ibrutinib的组合可显著提高对CLL细胞的细胞毒性[42]。TAK-659在LMP2A/Myc小鼠模型中以纳摩尔浓度抑制了脾肿大和肿瘤的发展,此外还阻断了肿瘤细胞向骨髓的转移。同时,TAK-659可以杀死肿瘤细胞而不损害宿主脾脏[43]。
在TAK-659的Ⅰ期临床研究中,10名年龄不小于18岁的晚期实体瘤或淋巴瘤患者(7例DLBCL,3例FL)接受TAK-659(60 ~ 120 mg,qd)口服治疗[44]。在7例可评估响应的DLBCL患者中,有3例患者实现PR。治疗过程中最常见的SAE是疲劳、谷草转氨酶(AST)升高、贫血和腹泻。8名患者(33%)出现3级以上的SAE。TAK-659口服剂量60 ~ 100 mg(qd)被认为可以开展进一步研究。在第一阶段人体研究的更新报告中,有54名晚期实体瘤/淋巴瘤成年患者在剂量递增期间(n= 35)接受了60、80、100和120 mg(qd)或在治疗期间剂量逐渐增加(n= 19)。其中24例可评估响应的DLBCL患者中,有11例(46%)达到了客观缓解(7例 CR,4 例PR);所有3例可评估响应的FL患者均达到PR,但是没有实体瘤患者达到客观缓解[45]。目前Ⅱ期临床评估TAK-659对R/R DLBCL患者的有效性和安全性研究已经被终止,原因是虽然无安全性隐患但缺乏足够有效性。
TAK-659治疗R/R AML的Ⅰb/Ⅱ期临床正在进行中。在Ⅰb期剂量递增期间,R/R AML的成人患者接受口服TAK-659剂量分别为60、100、120和160 mg(qd)。在药效动力学(PD)研究中,4名患者[2名 FLT3-ITD,2名 FLT3-WT(野生型)]用药后外周AML母细胞中核糖体蛋白S6在基线时被检测到并在之后降低,同时观察到FLT3-ITD磷酸化的抑制和周围成纤维细胞早期活性降低[46]。在最新报告中未观察到剂量限制性毒性(DLT)。最常见的SAE是31%的患者AST升高,23%的患者谷丙转氨酶升高和23%的患者淀粉酶水平升高。结果表明双重抑制Syk和FLT3的独特作用机制值得在具有FLT3突变的R/R AML患者中进行进一步研究。同时最新的研究证明,表达E3泛素连接酶胱硫醚β-裂合酶(cystathionine beta-lyase,CBL)突变体的髓样白血病对FLT3抑制剂高度敏感,并且可以通过抑制Syk激酶显著增强这种作用[47]。
3.7 其他
除了上述报道较多的Syk抑制剂,还有一些其他处于Ⅱ期临床的候选药物。MK-8457(8)是由默克公司研发的Syk抑制剂,对Syk的IC50为0.5 nmol · L-1,适应证为RA和高血压。多靶点激酶抑制剂TOP-1288(9),对Syk、MAPK、糖原合成酶激酶-3(glycogen synthase kinase-3,GSK-3) 的IC50分别为591、224和411 nmol · L-1。基于R406开发的JAK3/Syk双靶点抑制剂R348(10),具有良好的药代动力学性质和生物活性。其有效地减少了急性同种异体心脏移植排斥反应,适用于他克莫司的联合治疗[48]。
4 结语
Syk在BCR、FcR或黏附受体信号通路中具有关键作用,其被认为是治疗血液系统肿瘤、自身免疫性疾病的有效靶点。上市药物fostamatinib增强了人们对Syk抑制剂研发的信心,但存在低选择性引起的剂量毒性等问题。选择性高和耐受性好的第2代Syk抑制剂在血液肿瘤(主要为CLL)治疗中表现出良好的治疗前景,因此高活性和高选择性的Syk抑制剂还有待研究者继续开发。而针对RA等适应证临床试验的纷纷失利,人们开始思考是否仅抑制Syk不能产生足够的疗效,因此Syk抑制剂开发的一个新方向是双靶点抑制剂,如cerdulatinib为Syk/JAK双靶点抑制剂,其中JAK激酶在自身免疫性疾病中具有重要意义;TAK-659为Syk/FLT3双靶点抑制剂,FLT3对血液恶性肿瘤的发生发展有重要意义。同时组合疗法也是提升疗效的重要策略,例如Syk抑制剂与BTK、PI3Kδ等抑制剂的联用,其在临床上初步显示出较强的抑制肿瘤效果以及不易产生耐药性的优势,这对Syk抑制剂的进一步开发具有重要意义。
