ARSE基因突变导致X-连锁隐性遗传点状软骨发育不良的临床表型及基因型分析
2021-03-26李林杜开先张晓莉董燕关静甘玲贾天明
李林,杜开先,张晓莉,董燕,关静,甘玲,贾天明
X-连锁隐性遗传点状软骨发育不良(CDPX1,OMIM:302950)是一种罕见的先天性骨骼和软骨发育障碍性疾病,由位于染色体Xp22.3上的芳香基硫酸酯酶E(ARSE)基因[1]突变引起。该病首次由MAROTEAUX[2]报道,CDPX1的典型特征是软骨及骨骺的点状钙化、鼻和面中部发育不良、末节指骨发育不良,有些患者合并身材矮小、认知及运动发育障碍。该病临床表型轻重不一,本文报道1例新的ARSE基因突变所致CDPX1患者的临床表型和基因型,并结合既往文献总结CDPX1的临床表型及基因型。
1 临床资料
1.1 一般资料 患儿,男性,8个月19 d,患儿系第2胎第2产,胎儿四维超声提示鼻梁低平,额鼻夹角增大,两鼻孔间距增宽,上牙槽宽平。羊水染色体微阵列检查未见性染色体和1~22号染色体拷贝数异常,未见缺失、重复或大区域的杂合缺失,未见有明确临床意义的微缺失、微重复、杂合缺失。胎儿超声心动图提示动脉导管流速增快,右心室稍增大,心包积液。患儿于胎龄36+5周因“胎心慢”行剖宫产,出生时体质量为3.3 kg,新生儿Apgar评分为10分。出生后17 h出现呼吸困难后转入我院新生儿科,住院后测身长43 cm(低于同胎龄新生儿身长第3百分位[3]),胸部X线检查提示早产婴肺,左侧胸廓略塌陷;脊柱X线检查提示椎体、椎弓根形态异常,骨质密度不均,呈斑点状骨质沉积,椎弓根间距变窄(见图1);双下肢X线检查提示双侧股骨及胫腓骨骨皮质稍变薄,髓腔密度稍增高,双足诸骨密度减低,呈多发颗粒样骨质改变,腰骶椎椎体形态欠规整,呈多发颗粒样骨质改变(见图2)。心脏超声提示房间隔缺损(Ⅱ)2.5 mm、动脉导管未闭。听力筛查双耳未通过,眼底检查未见明显异常。给予经鼻持续正压通气(nCPAP)辅助通气3 d及对症治疗。
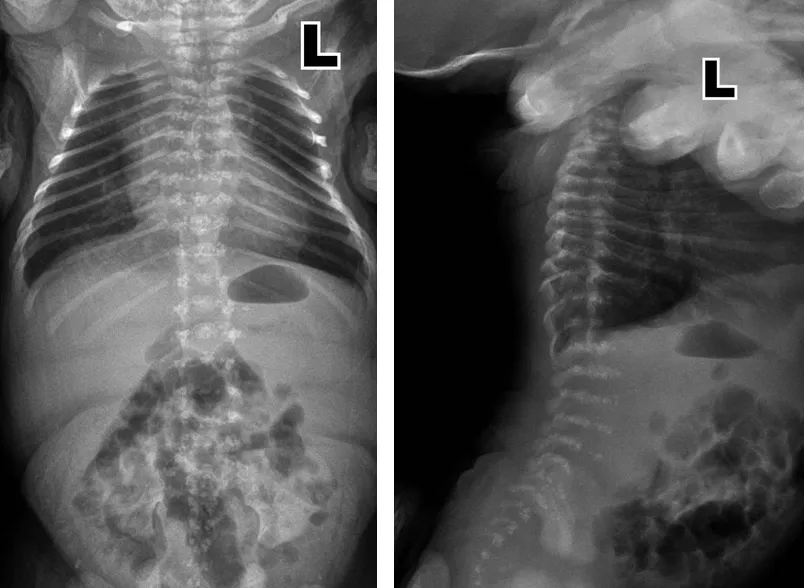
图1 出生后3 d脊柱正侧位X线检查Figure 1 Lateral X-ray of the spine of this case of CDPX1 caused by ARSE gene mutation on the 3rd day after his birth
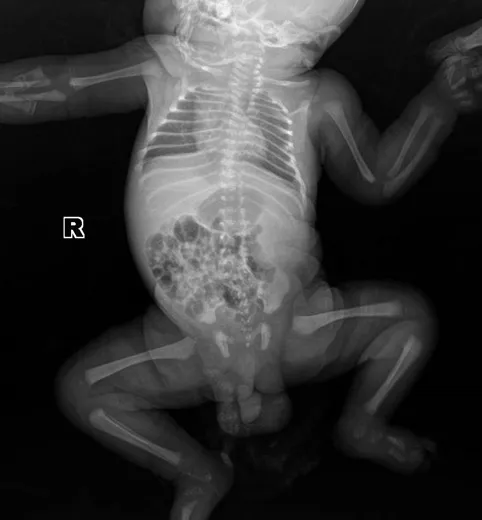
图2 出生后5 d双下肢正位X线检查Figure 2 Frontal X-ray of legs of this case of CDPX1 caused by ARSE gene mutation on the 5th day after his birth
患儿于4个月21 d时至我院复查,发现发育落后,表现为注意力明显不集中,不易逗笑,中线位置无明显竖头姿势,俯卧位抬头相当于新生儿水平,双手不会主动抓物,扶坐腰背无支撑。查听力检查提示混合性耳聋。颅脑核磁共振成像(MRI)提示脑外积液,双侧乳突积液,脑白质发育相当于4月龄,内耳水成像未见明显异常。发育评估提示认知及运动发育均落后。血串联质谱分析未见明显异常;尿代谢筛查提示尿嘧啶略高,未发现其他异常代谢产物。耳聋基因检测未发现最常见耳聋基因突变。查染色体核型分析:46,XY。给予康复治疗,期间建议患儿完善基因检测,家长拒绝。患儿8个月19 d时仍不能独坐,再次入院。
患儿父亲体健;母亲28岁,3年前确诊为系统性红斑狼疮(SLE),口服激素治疗,孕期仍口服小剂量激素,否认高血压、糖尿病等病史,否认孕期毒物、化学品及射线接触史,孕期规律产检;有1姐姐,5岁余,体健;否认家族中有类似疾病史及遗传病史。本研究通过郑州大学第三附属医院医学伦理委员会批准,患儿监护人对所做研究知情同意,并签署知情同意书。
1.2 体格检查 意识清,精神可;特殊面容:额部突出、双侧眼球突出、眼距宽、鼻梁低平、小下颌;营养中等,发育落后;全身皮肤黏膜无黄染、皮疹及出血点。前囟平软;双眼睑无水肿,结膜无充血,巩膜无黄染,双侧瞳孔等大等圆,对光反射灵敏;口唇红润,口腔黏膜光滑完整,咽无充血。颈软,颈短,无抵抗;气管居中;胸廓畸形,左侧胸廓略塌陷,吸气性三凹征阴性;双肺听诊呼吸音清,未闻及干湿性啰音。心音有力,律齐,心脏各瓣膜听诊区未闻及杂音。腹软,无压痛及反跳痛,肝右肋下1 cm,质软边锐,脾肋下未触及,肠鸣音正常。腰骶部脊柱向后凸出;四肢短小,手指粗短;肛门及外生殖器未见异常;生理反射存在,病理征阴性。专科查体注意力集中,能逗笑,会抓物,不能独坐,扶坐腰背支撑差,俯卧位抬头相当于2~3月龄。
1.3 影像学检查 脊柱X线检查:颈胸部交界处前突,T1~T8椎体及附件畸形,部分融合,六分腰椎,L2~L6椎体形态失常,椎体偏小,骶尾椎形态失常,提示脊柱多发复合畸形(见图3)。颅脑MRI:双侧额颞部蛛网膜下腔增宽,脑白质发育相当于7~8月龄,斜坡轻度凹陷(见图4)。脊柱MRI:胸椎曲度失常,颈胸段轻度侧弯,颈胸段至约T8椎体、腰骶椎部分椎体呈双面凹状,提示多发复合畸形(见图5)。
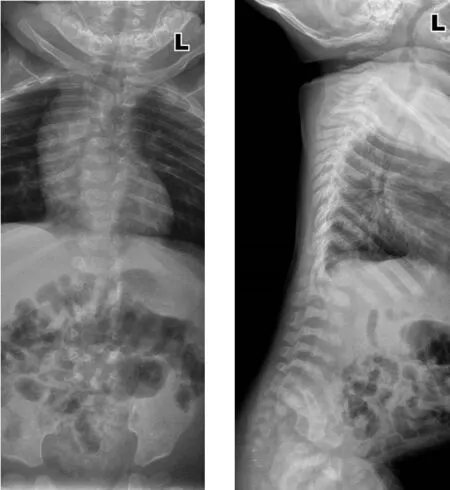
图3 8个月19 d脊柱正侧位X线检查Figure 3 Lateral X-ray of the spine of this case of CDPX1 caused by ARSE gene mutation at 8 months and 19 days
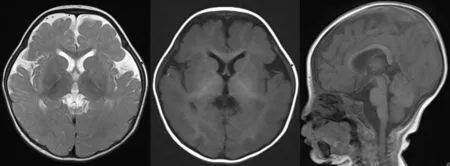
图4 8个月19 d颅脑MRIFigure 4 MRI of brain of this case of CDPX1 caused by ARSE gene mutation at 8 months and 19 days
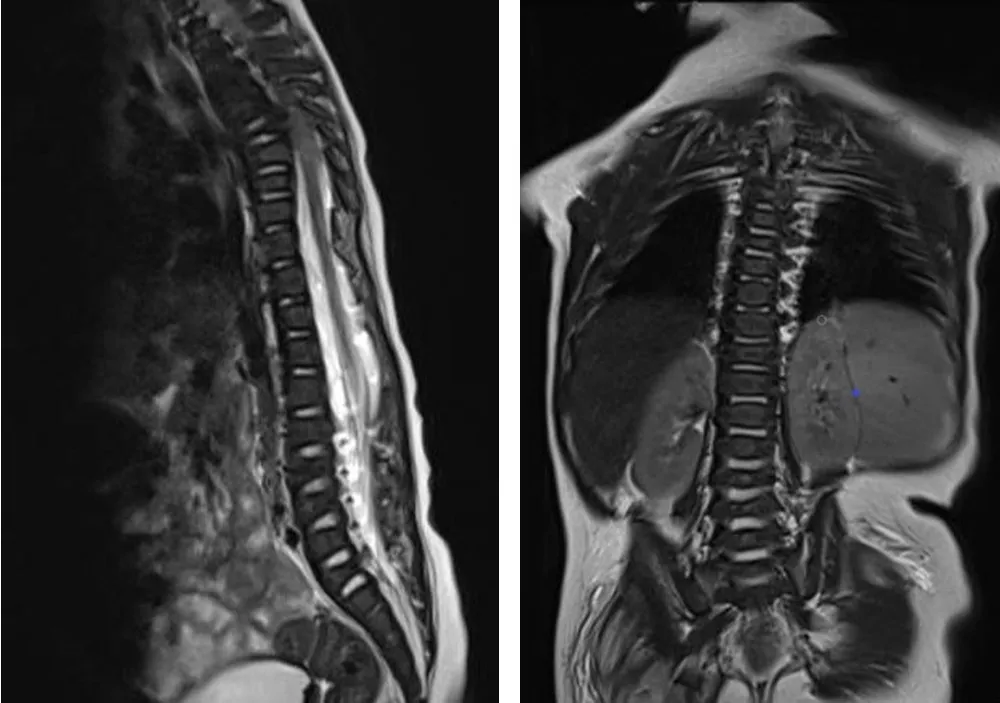
图5 8个月19 d胸椎及腰骶椎MRIFigure 5 MRI of thoracic and lumbosacral vertebra of this case of CDPX1 caused by ARSE gene mutation at 8 months and 19 days
1.4 全外显子测序检查 征得患儿父母知情同意后,抽取先证者、先证者姐姐及父母外周血各2 ml送至北京信诺百世医学检验所有限公司进行检查,结果示:先证者携带的ARSE基因(NM_000047.2)c.1219G>A(p.E407k)半合子变异,经Sanger验证遗传自受检者母亲,先证者母亲为杂合子变异,先证者姐姐和父亲未携带该变异(见图6),查询人类基因组突变数据库(HGMD)和Clinvar数据库,该变异国内外均未报道,未在正常人群中检出,生物信息分析软件SIFT、PolyPhen2和Mutation_Taster均预测有害。患儿临床症状与ARSE基因突变所致CDPX1高度相符,判定ARSE基因c.1219G>A变异致病。
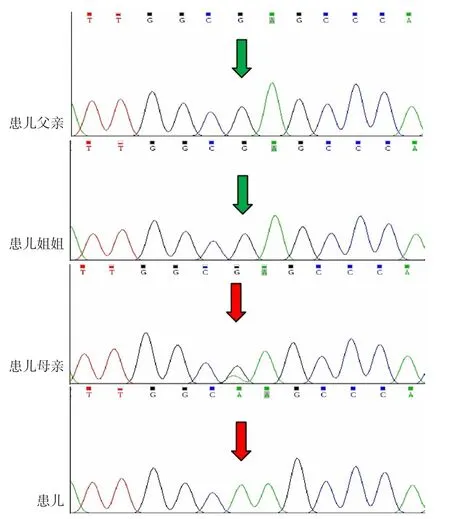
图6 全外显子测序及Sanger验证结果Figure 6 Results of whole exon sequencing and Sanger verification
1.5 随访 患儿一直进行康复治疗,2岁时可扶物侧行,可独站片刻,不能独走,精细动作及语言发育均明显落后于同龄儿童。复查脊柱正侧位X线:颈椎短,结构形态欠佳,颈胸部交界处前突,T1~T8椎体及附件畸形,部分融合,六分腰椎,L2~L6椎体形态失常,椎体偏小,L5以下椎板未融合,骶尾椎形态失常(见图7)。颅脑MRI:脑白质发育相当于24月龄(见图8)。全脊柱MRI:颈椎曲度变直,胸椎后凸,颈椎及上胸段椎体形态失常,椎间隙消失或明显变窄,部分椎体明显变扁或呈前后裂椎改变,下端胸椎及腰椎椎体前后径变小,椎体形态不规则,椎管内未见明显异常(见图9)。
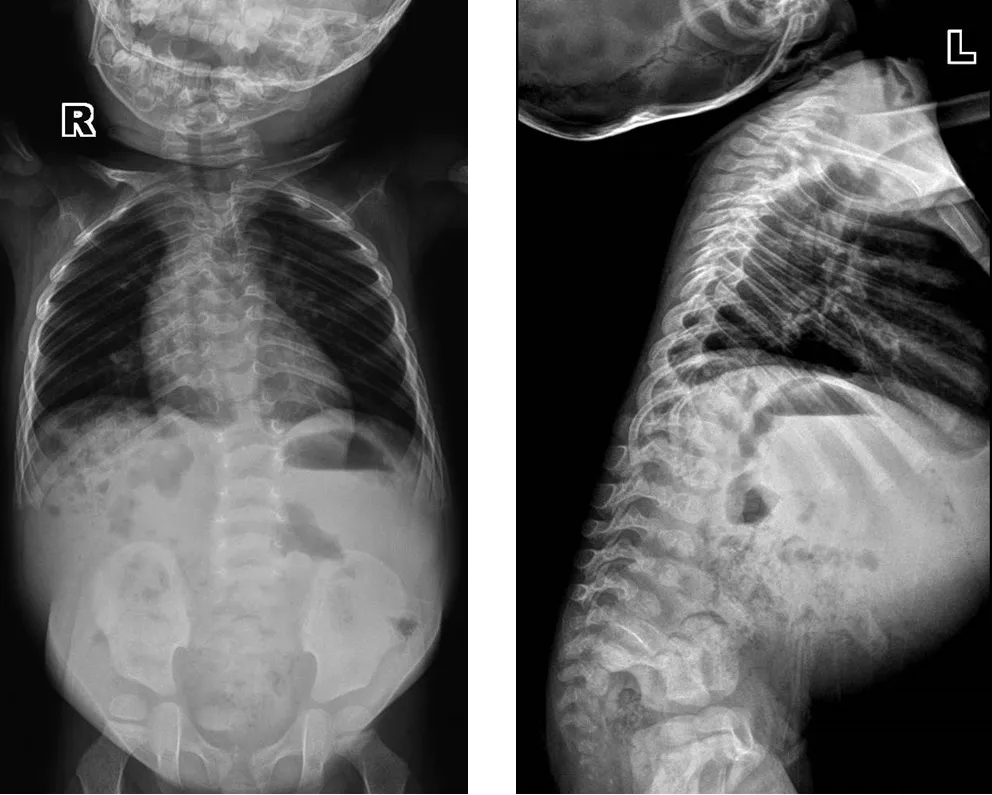
图7 2岁脊柱正侧位X线检查Figure 7 Lateral X-ray of the spine of this case of CDPX1 caused by ARSE gene mutation at 2 years old
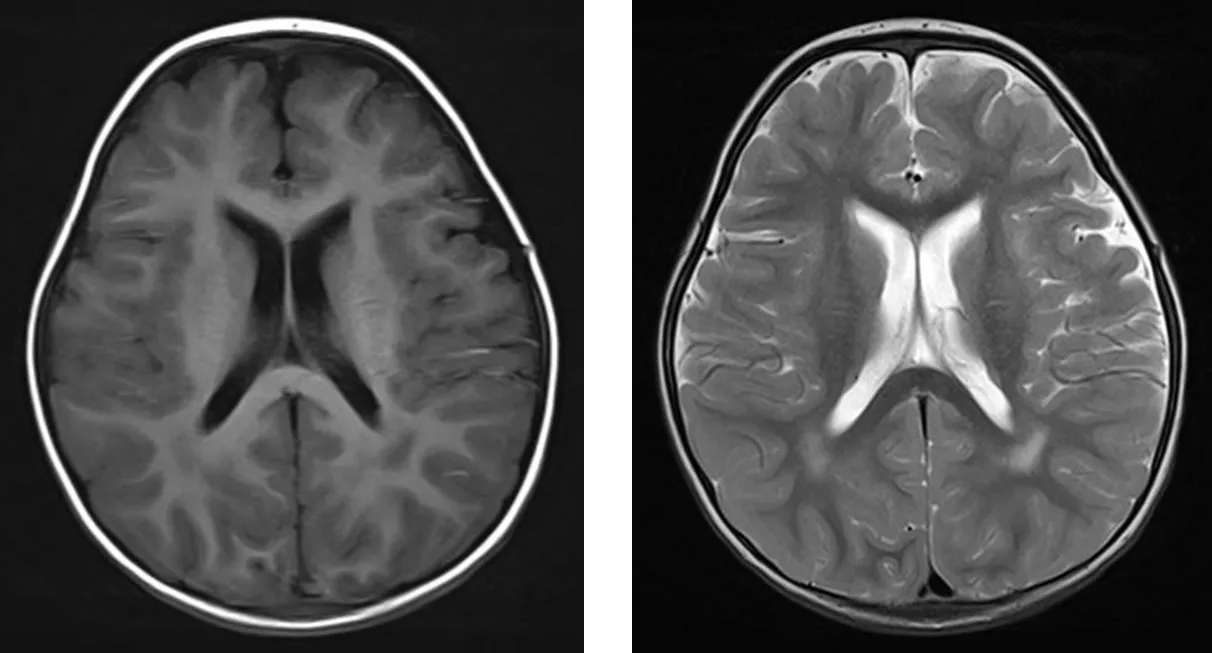
图8 2岁颅脑MRIFigure 8 MRI of brain of this case of CDPX1 caused by ARSE gene mutation at 2 years old
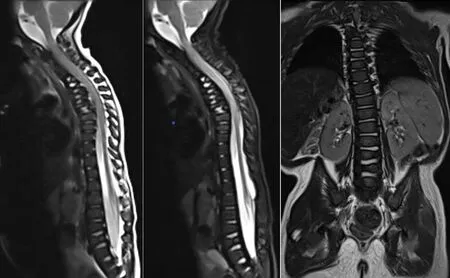
图9 2岁脊柱MRIFigure 9 MRI of the spine of this case of CDPX1 caused by ARSE gene mutation at 2 years old
2 文献复习
以“点状软骨发育不良”“芳香基硫酸酯酶E”和“Chondrodysplasia punctata”“ARSE”为检索词,分别在中国知网(CNKI)、万方数据知识服务平台、PubMed以及HGMD进行检索,筛选出存在ARSE基因变异且有详细临床表型的中文个案报告2例[4-5]及7篇相关英文文献[6-12],共23例记录了详细临床表型的患者。对包括本例共24例患者进行分析,其中男23例,女1例;胎儿期确诊4例,其中2例活产,1例26周流产,1例不详;余患者中1例因新生儿期合并弥散性血管内凝血(DIC)死亡。24例患儿中1例随访至20岁,身高184 cm;1例16岁时身高170 cm。临床表现:面中部发育不良(22/24)包括扁平脸,鼻尖扁平,鼻梁短、塌陷,鼻孔反曲,人中长,鼻翼与鼻尖之间有沟,耳位低;远端指骨短(14/24),脊柱及四肢长骨等点状骨骺(10/24),呼吸功能不全(8/24,其中反复发作的呼吸功能不全2例),听力异常(5/24),脊柱弯曲或椎体形态异常(5/24),鼻孔狭窄或闭锁(3/24),矮小(3/24),先天性心脏病(2/24),认知障碍(2/24),颈部短(2/24),气管及支气管钙化(2/24),气道狭窄(2/24),喉软骨钙化(1/24),肌张力低下(2/24),并趾畸形(2/24),体质量增长缓慢(2/24),喂养困难(2/24),宫内生长受限(1/24),小于胎龄儿(1/24),白内障(1/24),胸廓小(1/24),椎管狭窄(1/24),关节挛缩(1/24)。其中胎儿期表现为:面部扁平,不同程度的鼻发育不良,鼻梁凹陷,脊柱弯曲异常,椎体异常,椎管狭窄,上肢短,骨骺及脊柱弥漫性点状钙化,长骨发育不良,胎儿生长受限。检索HGMD,截至2020年2月共收录ARSE基因变异47个(致病性变异44个,可疑致病性变异3个):ARSE基因点突变37个(错义突变33个,移码突变2例,无义突变2个);ARSE基因单个或多个外显子缺失6个;ARSE基因完全缺失3个(包括ARSE基因的大片段缺失2个);ARSE基因的染色体易位1个。
3 讨论
点状软骨发育不良(CDP),1914年由CONRADI首先报道,国外统计发病率约为1/100 000活产婴儿[13]。CDP是以骨骺软骨不规则钙盐沉着为特征的一类骨发育异常疾病,临床和遗传上均有差异性。临床主要表现为肢体短缩,关节挛缩、髋关节脱位、多指、并指等畸形,有额部突出、眼距宽、鼻梁塌陷、外耳发育不良、高腭弓、腭裂、短颈等特殊面容,可伴有白内障、视神经萎缩或发育不良等眼部病变,部分有鱼鳞症、红皮症[14]等皮肤异常。根据遗传方式及临床表现,该病分为5型:(1)肢根型:常染色体隐性遗传,因多种过氧化物酶缺乏所致[15],多在1岁内死亡,预后最差;(2)非肢根型:常染色体显性遗传,发病机制不明确;(3)CDPX1型:X连锁隐性遗传,ARSE基因突变导致;(4)CDPX2:X连锁显性遗传,因Emopamil结合蛋白(EPB)基因突变[16]所致;(5)Sheffield型:发病机制及遗传方式尚不明确。本文报道患儿为ARSE基因半合子变异所致CDPX1。
CDPX1的致病基因是 ARSE基因,ARSE基因首次在1995年分离出来,位于Xp22.3处一组具有高度序列同源性的、逃避X失活、Y染色体上有伪基因的连续芳基硫酸酯酶基因内[1]。人类基因组中有17个硫酸酯酶基因,均具有广泛的序列同源性,并含有高度保守的半胱氨酸,该半胱氨酸经过独特的翻译后修饰,对硫酸酯酶的催化活性至关重要[17]。ARSE定位于高尔基膜[18],是一种耐热的芳基硫酸酯酶,可水解4-甲基伞形酮(4-MU)硫酸盐,因此体外利用4-MU硫酸盐测定ARSE酶活性,且酶活性在pH中性条件下最大[1,19]。目前ARSE的天然底物仍不清楚,既往报道ARSE活性可被华法林抑制[1],并且有文献报道妊娠早期应用华法林所致华法林胚胎病和妊娠期维生素K缺乏的胎儿[18]与CDPX1临床表型相似,但在这些患者中未发现ARSE基因突变[20-21],因此推测ARSE可能参与维生素K的代谢。至今发现的包括本例在内的ARSE基因突变中79%为点突变,且目前文献报道中接受ARSE基因检测的确诊患儿的母亲均为携带者[20,22],未发现新发突变的病例,本研究患儿ARSE基因突变亦是遗传自母亲,为首次报道的ARSE基因c.1219G>A突变。
CDPX1典型的临床特征:(1)点状骨骺:最少累及踝关节和末节指(趾)骨,也可累及长骨、椎骨、髋关节、肋骨软骨连接处、舌骨、气管和支气管软骨等。点状骨骺最早可在妊娠中期超声观察到,2~3岁时趋于改善或消失。(2)远端指(趾)骨短,X线检查可发现远端指(趾)骨短,呈倒三角形改变,指骨、掌骨和跖骨发育不良或者变宽。(3)鼻和面中部发育不良:鼻扁平,鼻尖塌陷,鼻梁短,鼻翼内有垂直沟槽。有文献表明鼻和面中部发育不良与患儿出生后呼吸窘迫相关[6]。(4)混合性耳聋。(5)身材矮小。另有文献报道19%~33%的患者出现运动发育迟缓[20];16%~20%的患者出现认知发育迟缓[22];20%的患者出现颈髓异常[20],且颈髓异常在儿童期可迅速加重[23],最终需外科治疗。CDPX1患者临床表型差异较大,一些患者仅有轻度面中部发育不良和远端指(趾)骨短[24],但也有一些患者存在严重并发症,包括颈椎体异常导致的脊髓受压,气管和支气管软骨广泛钙化导致的严重气道狭窄[20,25],甚至出现新生儿期死亡[11]。本研究检索到3例ARSE基因c.1743G>A突变,但3例患儿临床表型均不一致,其中1例仅有面中部发育不良和远端指骨短;1例气管和支气管的弥漫性钙化,出现反复发作和严重的呼吸困难,需机械通气。既往有文献分析了17例CDPX1患者的基因型和临床表型严重程度,发现不同基因型的患者有难以区分的临床表型,证实了ARSE突变基因型与疾病严重程度之间没有联系[22],而且同一家族中不同男性患者临床表型严重程度也有差异[5,7]。目前国内对CDPX1认识较少,本例为国内首次对CDPX1患儿从胎儿期到婴儿晚期完整的临床表型的报道,该患儿胎儿期有典型的鼻发育不良,新生儿期鼻梁低平、四肢短小、手指短、身材矮小、有典型的点状钙化和呼吸功能不全、椎体形态异常、听力异常,婴儿期发现认知和运动发育均落后。与以上文献报道不同的是,本例患儿8月余时椎体点状钙化消失,目前未发现脊髓病变,给予康复治疗后患儿认知运动发育均有进步,但仍落后于同龄正常儿。末次随访时患儿2岁,不能独走,可扶物侧行,可独站片刻,认知落后于同龄儿。
CDPX1预后差异较大,总体预后较好,大多数患儿症状较轻,智力和寿命正常,而且骨骼病变随年龄增长而减轻,但是严重的可能出现产前死亡、新生儿期死亡。产前超声或MRI检查可发现胎儿面中部发育不良,鼻梁小或缺失,额鼻角变大,方形的下牙槽嵴,鼻咽前部液体减少,椎体点状改变[3],椎管狭窄[6]。但随着孕周增加,软骨开始钙化,点状钙化可被正常钙化覆盖,给产前诊断带来困难。因此了解正常超声产前骨化中心出现的时间有助于检测出异常的点状改变[26],有助于疾病的早期诊断。该病无有效治疗方法,主要为临床对症治疗。对于存在鼻和面中部发育不良、软骨点状钙化、远端指骨短、身材矮小等的患儿应尽早完善染色体核型及高通量测序等遗传学检测明确病因,可为产前诊断和家庭遗传咨询提供依据。
作者贡献:李林、张晓莉、董燕、关静、甘玲进行案例收集和分析及文献检索;李林进行论文撰写及论文修订;杜开先、贾天明负责文章的质量控制及审校;杜开先对文章整体负责,监督管理。
本文无利益冲突。
