Multi-scale molecular dynamics simulations and applications on mechanosensitive proteins of integrins∗
2021-03-19Shouqin吕守芹QihanDing丁奇寒MingkunZhang张明焜andMianLong龙勉
Shouqin Lü(吕守芹), Qihan Ding(丁奇寒), Mingkun Zhang(张明焜),3, and Mian Long(龙勉),†
1Center of Biomechanics and Bioengineering,Key Laboratory of Microgravity(National Microgravity Laboratory),Beijing Key Laboratory of Engineered Construction and Mechanobiology,and CAS Center for Excellence in Complex System Mechanics,Institute of Mechanics,Chinese Academy of Sciences(CAS),Beijing 100190,China
2School of Engineering Science,University of Chinese Academy of Sciences,Beijing 101408,China
3Chongqing Engineering Research Center of High-Resolution and 3D Dynamic Imaging Technology,Chongqing Institute of Green and Intelligent Technology,Chinese Academy of Sciences,Chongqing 400714,China
Keywords: molecular dynamics simulations,mechanosensitive protein,allosteric dynamics,integrin
1. Introduction
Proteins are direct players for life,and corresponding biological function is dependent on their conformational features as well as dynamics properties. Many proteins present distinct functional conformations and the switching from one to the other,also known as allostery,lies on conformational dynamics as well as intramolecular interaction network. The initial definition of allostery is based on conformational change of the active site of a target protein induced by binding of an effector to this protein at another specific site, distinct from the active site.[1]Nowadays, the concept of allostery is extended to the phenomenon in which two sites on a single biological molecule are dynamically coupled despite being outside of direct physical interaction range. More and more proteins are known to possess allosteric features. For example,the extension of hinge region between EGF(epidermal growth factor) and Lectin domains of a cell adhesion molecule, selectin, induces the conformational change of epitope Lectin domain for better PSGL-1 (P-selectin glycoprotein ligand-1)ligand binding.[2-4]The K+ions conduction ability and kinetics of Kir(inward-rectifier potassium)channels depend on precise couplings among different gates through conformational regulations of all subunits.[5,6]Briefly, allostery is a key feature for most of proteins and plays pivotal roles for corresponding biological functions. Allosteric regulation has been widely employed in various fields from drug discovery to basic sciences.[7]Thus, investigations of allosteric behaviors and transition mechanisms are important for understanding structure-function relationship and functional modulation of proteins as well as the rationale of drug design. Furthermore, the driving force for protein allostery is its conformational dynamics, and the conformational dynamics are determined by intrinsic bonds and non-bond interaction network among all atoms. So high-resolution atomic-level evolution dynamics with time are essential for investigating protein allostery.
2. Introduction of molecular dynamics simulations
2.1. Biophysical principles of MDS
Experimental measurements, theoretical analyses, and numerical simulations are three main methods for scientific research.While x-ray crystallization,[8]NMR(nuclear magnetic resonance)[9]and Cyto-EM (Cryo-electron microscopy)[10]are main experimental methods for high-resolution structural biology,only static conformational features are offered by either x-ray or Cyto-EM, and limitations of NMR on molecular size and time resolution also hinder its application on protein allostery study. Other single-molecule experimental techniques, such as FRET (F¨orster resonance energy transfer),BFP(biomembrane force probe),AFM(atomic force microscopy), OT (optical tweezers), and MT (magnetic tweezers),could characterize allosteric features or dynamics of proteins in in vivo environment through indirect fluorescent or mechanical indicator,[11,12]but these methods could not offer visual features of atomic-level conformations with limited temporal or spatial resolutions. Molecular dynamics simulations (MDS) method was first developed in the late 1970s for elaborating the dynamics of a folded globular protein,[13]which is an ideal approach for studying conformational dynamics and allosteric pathways of proteins at atomic level. On one hand,computational simulations greatly save the expenses and extensive labors costed in experiments. On the other hand, the computational complexity of quantum-mechanical motions or chemical reactions is reduced in MDS since it is just based on Newtonian physics to simulate atomic motions.MDS is applied more and more extensively in various fields upon the rapid developments of computational ability(Fig.1).
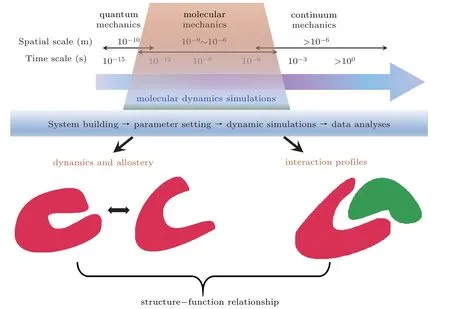
Fig.1. Roles of MDS in elucidating structure-function relationship of proteins. Conformational dynamics and allosteric pathways of single protein, or the interaction profiles of two paired proteins can be predicted from MDS at the specified spatiotemporal resolutions. The main procedures for running MDS were also profiled.
The principle of MDS is that each atom of simulation system is treated as a basic particle, and classical Newton’s second law(Eq.1)serves as the governing equation,
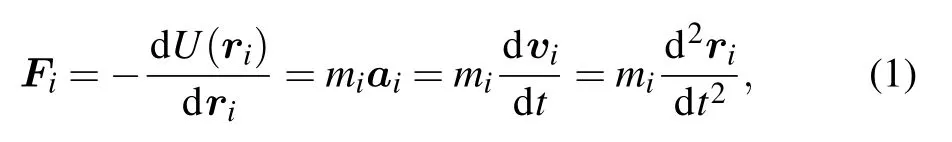
where Fiand U(ri)are the force and energy acting on the particle i,respectively. ri,mi,ai,viare the position,mass,accelerated velocity,and velocity,respectively. t is the time. A trajectory that features the evolutions of positions and velocities of particles with time is obtained by solving the differential equation Eq. (1). The prerequisite for MDS includes the initial conformation of target protein(ri),particle type(mi),and interaction potential U(ri)between any two particles.The initial conformation can be obtained from x-ray crystallographic,NMR,Cyto-EM or homology-modeling methods.The particle type depends on the atomic type. For biomolecules,empirical potential fields are usually used for describing the interaction energy between atoms as follows:[14]
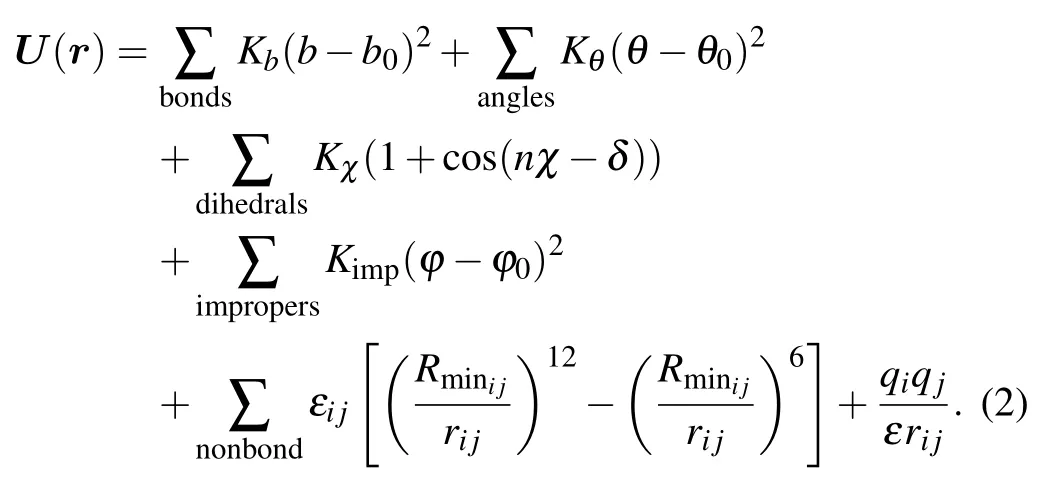
These interaction energy includes bond and non-bond interactions.The former contains bond stretching energy,angle bending energy, dihedral angle torsion energy, and non-coplanar improper energy corresponding to the 1st to the 4th terms of the right-hand side of Eq.(2),respectively. The last two terms are non-bond interactions of van der Waals interactions represented by the Lennard-Jones potential and electrostatic interactions quantified by Coulomb’s law. Corresponding parameters include bond length b (equilibration length, b0), bond angle θ (equilibration angle, θ0), dihedral angle χ, dihedral angle period n and phase angle δ,non-planer torsion angle ϕ(equilibration torsion angle,ϕ0). Respective spring constants are Kb,Kθ,Kχ,and Kimp. rijis the distance between particles i and j, εijis the corresponding energy barrier when the distance between two particles are Rminij, ε and q are dielectric constant and particle charge,respectively. Collectively,equation(2)with a set of these parameters is called a‘force field’because these parameters describe the contributions of various atomic-level forces that govern molecular dynamics. Several force fields are commonly used in molecular dynamics simulations, including AMBER (Assisted Model Building with Energy Refinement),[15]CHARMM (Chemistry at HARvard Macromolecular Mechanics),[16]and GROMOS (GROnigen MOlecular Simulation).[17]These force fields differ principally in the way they are parameterized but generally give similar results.
In general,the main procedures for running MDS include four steps, e.g., system building for defining the initial position of each atom and interaction modes among atoms, simulation parameter setting,dynamic simulation,and data analyses (Fig.1). Once the forces acting on each of atoms have been calculated, the positions of these atoms are moved according to Newton’s laws of motion in the dynamic simulation procedure. Many popular molecular dynamics simulation software packages are used extensively for investigating conformational dynamics of biological macromolecules, i.e.,AMBER,[18]CHARMM,[19]GROMACS,[20]and NAMD,[21]some of which bear the same names as their default force fields. The key contribution of MDS is to explain experimental phenomenon at microstructural levels or to predict conformational features and offer guidelines for experimental design. And a number of studies validate the effectiveness of MDS based on the consistency between the simulations and experimental data. For example, conformational dynamics differences resulted from site mutation upon MDS successfully offer the microstructural bases for understanding the impacts of these mutations on the gating dynamics of Kir channels and water permeability of rice plasma membrane intrinsic proteins.[5,22]Forced dissociation simulations of P-selectin-PSGL-1 or TCR-pMHC interactions demonstrate corresponding micro-structural mechanisms of their non-intuitive catch bond behaviors.[23,24]
2.2. Progress update for improving MDS
MDS is a powerful tool to study the structure-function relationship of proteins, focusing on conformational dynamics and allosteric behaviors derived by non-covalent interactions among atoms or particles but not configurational changes derived by formation or breakage of covalent bonds. However,conventional MDS for proteins or other biomolecules is time-consuming because of the interaction complexity among atoms,small integral time step of femtosecond(fs)that is comparable to atomic vibration, and large size of simulation system that includes not only target proteins but also water or membrane environment. Thus, adequate sampling of conformational states and elaborative observation of allosteric dynamics with conventional MDS remains difficult due to the necessary timescales, even upon the increasing computing power depending on hardware and software developments. In fact, above insufficiencies of the conventional MDS are contradictory because the atomic interaction needs to promote calculation accuracy while the adequate sampling needs to speed up the computation. To address this issue, two kinds of schemes are developed for satisfying respective requirements. The MDS based on more accurate empirical potential fields such as CFF(consistent force field),MMFF(the Merck molecular force field),and the combinations of MDS and QM(quantum mechanics)simulations are typical methods for improving calculation accuracy.[25,26]
By contrast, the strategies for promoting computational efficiency are multiple. One kind is the coarse-grained MD(CG-MD)simulation that allows longer time and larger spatial scale simulations through simplifying the simulation system.The basic idea of CG-MD simulation is to describe the biological macromolecule as a chain composed of coarse particles,which integrate basic units such as amino acids by ignoring atomic-level details, linked by different flexible springs. The calculation ability is accordingly speeded up due to the reduced particle number,reduced degree of freedom,and simplified interaction potential among particles.[27]It is worth mentioning that reactive CG-MD simulation method is also developed for considering both chemical reaction and computational ability.[28]In fact,the ultimate goal of MDS is to effectively search all the minimal or minimum energy surfaces of the molecular system for understanding its structural characteristics. Therefore, enhancing the sampling efficiency by artificially biasing potential barriers is another kind of methods.Based on this, accelerated molecular dynamics (AMD) have been put forward one after another. A typical representative is metadynamics[29]and its basic idea is to fill sand using corresponding deflection potential energy for each state to reach a nearly flat potential energy surface. Then the free energy surface of the system is the opposite deflection energy surface applied at each state. It should be mentioned that free energy is still difficult to be quantified upon MDS for flexible macromolecules that have minimum energy conformations separated by low-energy barriers, because the MDS cannot adequately sample phase spaces that make important contributions to the free energy. So accurate and effective calculations of free energy for macromolecules are still a challenge by now.
In addition,exerting external interference for speeding up simulations to achieve special biological process in acceptable timescale is another strategy for expanding applications of MDS. SMD (steered molecular dynamics)[30]and TMD(targeted molecular dynamics)[31]are the most representatives along this line.Initial implementation of the SMD method is to force the center of mass of the tagged atoms to move with constant velocity or constant force along a direction. The way of force loading is now extended at any time with variable force including magnitude and direction. The SMD method has been used widely for investigating conformational dynamics of forced unfolding of biomolecules or dissociation of molecular complex,such as unfolding simulations of titin modulus[32]and dissociation simulations of the biotin-avidin complex.[33]It should be noted that external force applied in SMD simulations is not only for speeding up simulations,but it also mimics physiological mechanical environment as described in the next section. The TMD method is to guide subset of atoms towards a final target structure by means of external forces. Unlike the SMD method, the external force of TMD is adjusted at any time based on the difference between the current and target structures. Firstly, the RMSD (root-mean-square deviation)between the current coordinates and the target structure is calculated at each timestep and the force on each atom is then gotten by the gradient of the potential,
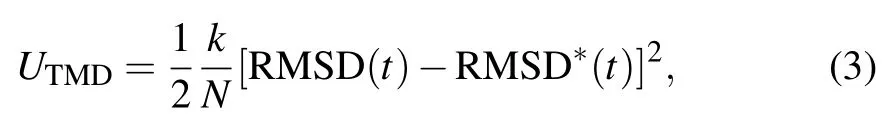
where RMSD(t)is the instantaneous best-fit RMSD of the current coordinates from the target coordinates, and RMSD∗(t)reduces linearly from the initial RMSD at the first TMD step to the final RMSD(generally set as 0)at the last TMD step. The spring constant k is scaled down by the number N of targeted atoms. The TMD method is widely used for investigating allosteric dynamics of biomolecules, as seen in identifying the conformational transition pathway of Kir channels upon the binding of PIP2(phosphatidylinositol 4,5-bisphosphate).[34]
In brief, applications of MD simulations are extended widely following the bringing forward of novel simulation ideas as well as quick developments of computational ability and algorithms. In addition,serial auxiliary tools are also developed effectively for building more complicated biomolecular simulation systems and for constructing corresponding force field parameters of unconventional elements,which undoubtedly expand the applications of MDS for more and more biomolecular systems. Nowadays, the major MDS software packages of NAMD, GROMACS, AMBER, and CHARMM are all improved greatly with integrating of QM/MM, CGMD, and SMD modules. Specifically for biological macromolecules,NAMD is favored with perfect SMD package and high extensibility based on flexible TCL(Tool Command Language) interface, and GROMACS is featured with its high computing efficiency.
3. Biological importance of mechanosensitive proteins
Life is a complex mechanical system including the omnipresent gravitational, osmotic forces as well as mechanical shearing, stretching, bending, twisting, compressing.[35]Such mechanical factors highly modulate life from immunobiology,[36]developmental biology[37,38]to genetic biology.[39]Unravelling corresponding mechanisms and implications requires the understanding of mechanical sensation,transmission,and transduction at cellular level.As a collection of protein machines acting in an elaborate interaction network,cellular responses for mechanical environments lie on mechnochemical properties of interacting proteins. For example,cells exert and transmit forces largely through polymerization and contraction of various cytoskeleton proteins and cooperation of corresponding adaptor proteins.[40]Typical receptors at the cell membrane, such as mechanosensitive ion channels PIZEO, G protein-coupled receptors (GPCRs), and integrins,can sense external mechanical environment indirectly through binding to their ligands or directly through deformation of the membrane itself. They can also transmit mechanical signals to cytoplasm through indirect or direct interacting with force-sensing elements of microtubules and actin filaments,bridging the linkage from extracellular mechanical microenvironment to cytoplasmic even to intranuclear biochemical signaling.[41-43]As a result, the conformational or allosteric dynamics of these membrane receptors upon mechanical regulations play key role on mechanical sensing, transmitting and transducing inside a cell. As an example, allosteric analyses were extensively discussed below for a typical cellular adhesion molecule,integrin.
4. Applications of MDS in mechanosensitive integrins
Integrins are heterodimeric type-I transmembrane (TM)proteins that mediate cell-cell and cell-matrix adhesions.[44]There are 24 members of integrin family assembled by 18 α subunits and 8 β subunits, and each β subunit contains 8 extracellular domains of I-like (βI or βA in brief), hybrid,plexinsemaphorin-integrin(PSI),EGF1-4,and β-tail domain.All the 18 α subunits contain 4 extracellular domains of βpropeller, Thigh, Calf-1, and Calf-2, and half of them have an additional I domain (αI or αA in brief) with its N- and C-terminals inserted into the β-propeller domain (Fig.2(a),left). The overall shape of an integrin ectodomain displays a large head supported by two long legs. Integrins adopt three distinct conformations by coordinating the arrangements of α and β subunits and conformational changes of intrasubunit domains, presenting inactive (bent-closed), intermediate (extended-closed), and active states (extended-open)(Fig.2(a), right). And the switching among different conformational states empowers integrins various ligand binding affinities and bidirectional signal transduction of insideout and outside-in signaling between extracellular microenvironment and intracellular activity for cellular adhesion and migration[44,45](Fig.2(b)). Thus, the allosteric dynamics of integrins are key to understand and elucidate their biological functions. While static structural features upon structural biology experiments[46,47]and indirect implications of conformational dynamics at molecular level upon single-molecule experiments[12,48]hinder allosteric dynamics of integrins due to the experimental limitations in temporal or spatial resolution, studies upon MDS make up the deficiencies and deep the understanding on microstructural dynamics of integrins.In addition, since integrin’s microstructural dynamics can be regulated by external force through binding to their respective ligands for mediating intercellular or cell-substrate adhesions,the force-induced conformational or allosteric dynamics are another important aspect for understanding their structurefunction relationship.
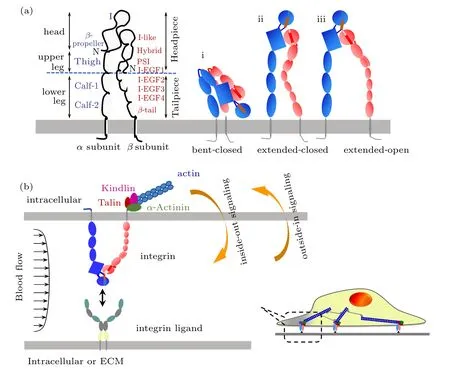
Fig.2. Conformational features(a)and corresponding biological signaling(b)of integrins. (a)Three conformations of bent-closed i,extendedclosed ii,and extended-open iii are proposed for distinct integrin members. (b)Two types of inside-out signaling and outside-in signaling are applied when a cell adheres to another cell or extracellular matrix(ECM)via integrin-ligand bonds.
A number of simulation studies on integrin dynamics range from all-atom simulations for understanding integrin activation at the level of individual molecules, to lower resolution coarse-grained, lattice-based, diffusion-reaction algorithms,and theoretical models for multiple integrins. The focuses of these studies cover the detailed conformational dynamics of a local domain to rough allosteric mode of a global molecule, as well as the ligand-induced conformational allostery with or without external force. For the I domaincontaining integrins, the αI domain not only serves as the binding pocket of external ligand based on its MIDAS(metal ion-dependent adhesion site)but it also binds to the pocket of βI domain as an internal ligand,[49]implying that the conformational and allosteric dynamics of αI domain play key role for integrin activation. In combination of SMD simulations and mathematical model,three distinct conformations are successively transitioned through pulling the C-terminus of its α7-helix for the αLβ2αI domain,and the coupling between these conformational changes of αI domain and its dissociation from ICAM-1 ligand under force is modeled for estimating the force-dependent kinetic rates of interstate transition.[50]Detailed conformational transfer from forced αI domain α7-helix C-terminal to the β6-α7loop and further to MIDAS loops and metal coordination are also investigated based on SMD simulations,and the key roles of specific ratchet residues for regulating αI domain conformational equilibria of intermediate state are evaluated using several I domain-containing integrins.[51]Also, SMD simulations are performed for comparing conformational stability of αLβ2and αMβ2αI domains in different affinity states and relevant I domain-ICAM-1 interaction features for elucidating their distinct biological functions. The unstable feature of αLβ2αI domain with diverse orientations of its α7-helix and spontaneous transition from low affinity state to intermediate affinity state is consistent with its versatile function in inflammation cascade than that of αMβ2.[52]Furthermore, two conservative salt bridge interaction pairs that constrain both the upper and bottom ends of the β2integrin αI domain α7-helix are determined for regulating α7-helix allostery through SMD simulations and experimental validation. The results indicate that the magnitude of the salt bridge interaction is related to the stability of the αI domain α7-helix and the strength of the corresponding force-induced allostery.[53]
Extracellular domains of integrin β subunit play pivot role for both outside-in and inside-out signal transmission of integrins. The conformational dynamics of the I-like and hybrid domains from the β3integrin are studied by MDS and normal mode analysis,and the results demonstrate the key role of I-like domain α1and α7helixes for the interdomain conformational transition from I-like domain to hybrid domain.[54]TMD simulations of the headpieces of both αIIbβ3and αvβ3integrins further confirm above observations, with the results that hybrid domain swing-out is accompanied by conformational changes within the I-like domain that propagate through the downward motion of α7-helix C-terminus to the opening of the β6-α7loop, which further induce conformational changes of I-like domain.[55]Taken together,these results suggest that intradomain and interdomain interactions are both responsible for β3integrin activation. Simulations of FnII(10)-bound αvβ3integrin headpieces are also carried out to identify the spontaneous or force-accelerated allosteric pathway along which ligand-induced strain propagates via elastic distortions of the I-like domain α1-helix to the I-like/hybrid domain hinge through a hydrophobic T-junction between the middle of the α1-helix and top of the α7-helix.[56,57]The effect of single amino acid site mutation on conformational dynamics of αIIbβ3indicates that L33V mainly displaces the equilibrium between common structures of β3subunit but not allostery.[58]And the importance of P163S mutation of β subunit happened in Glanzmann thrombasthenia disease is validated through exhibiting the distinct regulation of this site on the inter-subunit interactions of αIIbβ3and αvβ3based on MDS.[59]Similar study is also performed for investigating the importance of disulfide bond disruption formed by β3subunit C560 on αIIbβ3activation.[60]
Dimerization of TM helices of α and β subunits also plays a key role in integrin signaling. Multiscale simulations combining CG-MD and atomistic MD simulations are used to investigate the effect of specific site mutation of αIIbβ3integrin on the dimerization of TM helices, indicating that the dimer interface of αIIbβ3TM helices is more flexible for effective transbilayer signaling.[61]The self-assembly dynamics difference of α and β subunit TM helices between αLβ2and αIIbβ3integrins are also investigated using CG-MD simulations,suggesting that,due to the existence of specific interhelix hydrogen bond, the αLβ2TM helices packing is close-tooptimal with a deeper minimum free energy profile than that αIIbβ3.[62]
Both the starting of integrin inside-out signaling and transmitting of outside-in signaling from extracellular to intracellular depend on its intracellular complex between the integrin β subunit cytoplasmic tail,cell membrane and relevant cytoskeleton proteins. Through direct binding to both cytoplasmic tail of integrin β subunit and actin cytoskeleton,talin plays crucial role for integrin bidirectional signal transduction by bridging the extracellular matrix(ECM)or counterpart ligands and cytoskeleton (Fig.2(b)). A series of MD simulations are performed for investigating the effect of talin on integrin activation.Based on multiscale MDS,the features of talin head domains-cellular membrane interaction,the key residues of talin head domains for talin binding to membrane, and the micro-structural mechanism of talin head domains-induced activation of αIIbβ3are studied based on the simulation systems including talin head domains,cellular membrane and integrin TM and cytoplasmic tail domains. The results demonstrate that talin F2 and F3 domains binding re-orientate TM domain of β3subunit through its binding to negatively charged lipid headgroups in the membrane,and the perturbed interaction of talin to the membrane in the F2 domain mutant in turn perturbs the talin/integrin interactions.[63-66]Furthermore, models of the complete integrin receptor in complex with talin F2-F3 domain inserted in biologically relevant bilayers is constructed for studying the dynamics of the integrin receptor and its effect on bilayer structure and dynamics.[67]
In addition to above simulation studies that focus on local conformational dynamics or allostery of specific domain or interdomain hinge,global allosteric features undergoing large conformational changes of full-length integrin are also investigated by various simulation methods. All-atomic SMD simulations are used to investigate forced-unbending dynamics of a complete integrin αVβ3ectodomains in both un-ligated and ligated forms. And the results show that external force can activate integrins readily from bent to extended state through pulling the head of the integrin or a cyclic RGD ligand bound to the integrin. The major energy barrier along the unbending pathway is resulted from the interface interactions between the hybrid and both β subunit tail and EGF4 domains.[68]Allosteric dynamics consistent with the switchblade model followed by a separation of the TM helices is visualized for fulllength integrin αVβ3with TM helical and cytoplasmic tails based on the combination of all-atom equilibration and SMD simulations.[69]Similar simulations are also performed to investigate the activation characteristics by systems of RGDbound, full-length αIIbβ3with talin bound cytoplasmic and transmembrane domains. The results demonstrate the key role of talin binding on the separation of the integrin’s α and β subunits,as well as the activation pathway of integrin by RGD ligand via disrupting the key interaction group between I-like domain and β subunit tail domain.[70]Based on a modified CG heterogeneous elastic network model that can distinguish local fluctuations from global motions, the simulations of a full-length integrin αVβ3with TM helixes and cytoplasmic tails support the notion that the integrin extension can result from the disruption of weak, long-range interactions while maintaining structural connectivity through the short-rang and stronger connections. Meanwhle, the allostery of integrin is consistent with both deadbolt and switchblade mechanisms without excluding or strongly supporting either one.[71]A CG computational model is also developed for assessing the intrinsic mechanism of integrin-mediated adhesion assembly,as well as the role of actin filament architecture on the ability of integrin clustering and ligand binding.[72]
In brief, various MD simulations methods are applied widely for investigating allosteric dynamics of integrins,and these studies provide new insights into the structural mechanisms of integrin activation through revealing the dynamic processes ranging from local conformational fluctuations to global conformational dynamics and allosteric pathways.However,these studies mainly focus on specific event of integrin allostery that far from full view of intergrin activation.It is well known that typical conformational changes of integrin activation include extending of integrin headpiece,swing out of hybrid domain and separation of legs,and the existence of I domain for the I domain-containing integrins induces more complexity with the internal ligand binding. Is there any optimal happening sequence for these conformational changes during integrin activation? Apart from the extra internal ligand binding,is there any difference of conformational changes for activation between I domain-absent and I domain-present integrins? Is the allosteric pathway of integrin inside-out activation completely independent to that of outside-in activation or not? Addressing these questions need deep investigations evidently. In addition,as a well-established mechanosensitive receptor,the role of external force on integrin allostery is also far from being acknowledged.
5. Conclusions and perspectives
Structure determines function. Studies focusing on atomic-level micro-structural features of proteins provide the bases for understanding or regulating corresponding structurefunction relationship. More and more proteins are found to adopt different conformational states for implementing distinct biological functions. Intrinsic conformational dynamics with or without external factor regulation are the driving force of conformational allostery from one state to the other, and the characteristics of conformational dynamics or allosteric dynamics are intrinsic properties of proteins. Based on the progress of computer technologies and algorithms and the appearances of increasing high-resolution protein structures,MD simulations become more and more important for studying the structure-function relationship of proteins. On one hand,equivalent to theoretical analyses and experimental measurements,MDS is one of indispensable methods and used widely for predicting or explaining experimental behavior. On the other hand, current experimental technologies are limited in temporal or spatial resolution, and MDS has its unique advantage for deeply exploring the conformational or allosteric dynamics of protein. In fact, while basic structural features and overall allosteric models are proposed for those proteins based on experimental behaviors,detailed conformational and allosteric dynamics and corresponding micro-structural mechanism are far from clear. Thus, MDS approach has its irreplaceability for exploring micro-structural mechanism of target proteins by now.
While the rapid development and successful application on elucidating conformational dynamics of proteins,the MDS method still faces challenges based on the varied goals. First of all,there still is significant gap between available MD simulation timescale for proteins(ns ~µs)and real timescale(ms~s)of physiological processes by current computational ability. This gap is resulted from protein complexity that limits the setting of simulation timestep around fs and determines the complicate force field (Eq. (2), and inclusion of semiphysiological liquid or membrane environment increases particle number of simulation system sharply. Further speeding up of MDS is still a pressing desire. Secondly, protein complexity is represented by various modification including glycosylation, sulfation, phosphorylation, and so on. However,the effect of these modification on molecular function is usually neglected in MD simulations because of the lack of corresponding force field parameters.Further improvement of force field parameters for uncommon elements is necessary to predict conformational dynamics of target proteins more accurately. Lastly, exact mimicking of physiologically mechanical or physical microenvironment in MDS is crucial for investigating the structure-function relationship of mechanosensitive proteins such as integrins. Current simulations often simplified these regulation factors through idealized setting, i.e.,shear stress is simplified as a point pulling force. To a certain extent,these simplifications result in evaluation deviation of these regulation factors,and developing flexible module in MDS procedures for accurate mimicking of physiological regulation environment is required. In brief, the developing and popularizing of MDS are always complementary with the deep understanding of target proteins.
杂志排行

Chinese Physics B的其它文章
- Transport property of inhomogeneous strained graphene∗
- Beam steering characteristics in high-power quantum-cascade lasers emitting at ~4.6µm∗
- Enhanced spin-orbit torque efficiency in Pt100−xNix alloy based magnetic bilayer∗
- Soliton interactions and asymptotic state analysis in a discrete nonlocal nonlinear self-dual network equation of reverse-space type∗
- Discontinuous event-trigger scheme for global stabilization of state-dependent switching neural networks with communication delay∗
- Model predictive inverse method for recovering boundary conditions of two-dimensional ablation∗