中枢神经系统非典型畸胎瘤样/横纹肌样瘤10例临床病理分析
2021-03-17罗柏花龚光辉胡忠良李青玲
罗柏花,龚光辉,胡忠良,李青玲
非典型畸胎瘤样/横纹肌样瘤(atypical teratoid/rhabdoid tumor, AT/RT)是一种少见的中枢神经系统(central nervous system, CNS)恶性肿瘤,常见于婴幼儿、儿童,成人罕见,具有高度侵袭性,临床进展快,组织学形态复杂多样,易误诊为其他类型的肿瘤。本文收集10例AT/RT,探讨其临床病理学特征、诊断、鉴别诊断及预后等,旨在提高对其的认识水平。
1 材料与方法
1.1 材料收集2016年~2019年中南大学湘雅医院诊治的10例AT/RT,搜集其临床病理资料,主要包括患者一般资料、发病部位、临床病史、影像学资料及治疗预后随访等。根据WHO(2016)中枢神经系统肿瘤分类标准[1],对所有病例进行病理切片复查。
1.2 方法标本均经10%中性福尔马林固定,常规取材、脱水、透明、浸蜡、切片和HE染色。免疫组化染色采用EnVision法,一抗包括GFAP(MX047)、Olig2(EP112)、EMA(757F5D6)、p53(D07)、H3K27M(RM192)、Syn(214A4G5)、CD34(QBEnd/10)、INI1(MRQ-27)、CD99(PCB1)、vimentin(MX034)、Ki-67(MX006)等,抗体及免疫组化试剂购自福州迈新公司、北京中杉金桥公司和苏州百道医疗公司。实验步骤严格参照试剂盒说明书,染色过程中设阴阳性对照,阳性对照是用已知阳性组织切片,阴性对照用PBS代替一抗。
1.3 结果判定采用Barnes半定量法阳性判断标准,阳性结果呈棕黄色或棕色颗粒,由两位主治医师以上的病理医师对免疫组化染色结果进行双盲法阅片。
2 结果
2.1 临床特点10例AT/RT患者发病年龄1~6岁,中位年龄2.5岁,临床以呕吐、行走不稳为主要症状。肿瘤发生部位:小脑2例、脑室5例、额顶叶3例。肿瘤大小各异,MRI检查示肿瘤呈占位性病变,部分囊性变伴出血,增强扫描后均有不规则或不均匀强化(图1、2),其中5例伴有不同程度脑积水改变。10例患者均行肿瘤完整切除术。7例生存时间为1~15个月,中位生存时间为5.8个月,3例失访(表1)。
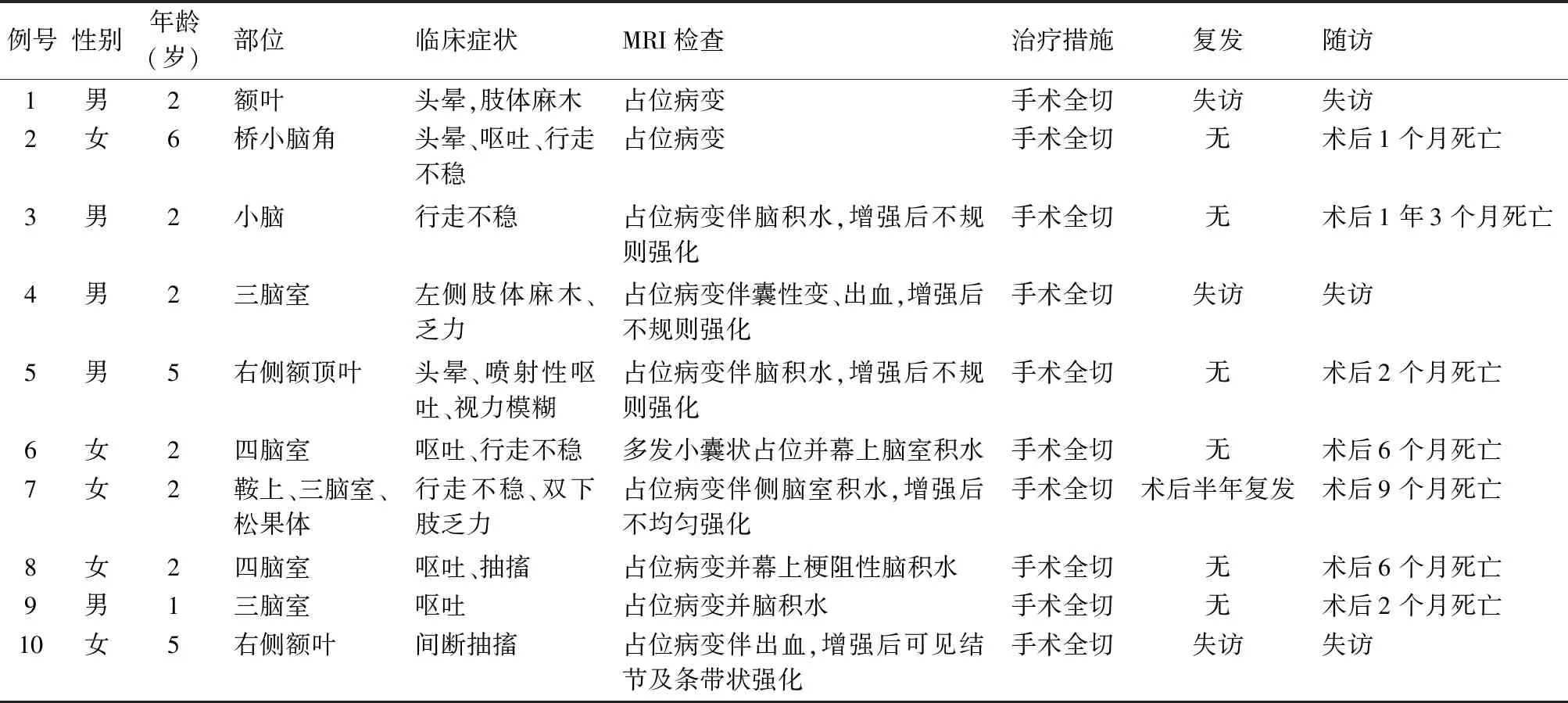
表1 10例AT/RT患者临床及影像学资料

图1 头颅MRI增强扫描:占位伴出血,增强后可见结节状强化 图2 头颅MRI增强扫描:混杂信号,不均匀强化
2.2 病理检查
2.2.1眼观 10例AT/RT患者行开颅脑肿瘤切除术,肿瘤呈浸润性生长,为边界欠清的灰白、灰红色肿块,2例呈囊性变伴出血,2例有胶冻状、半透明状。
2.2.2镜检 镜下见肿瘤细胞弥漫成片,肿瘤细胞呈多形性,有梭形细胞(图3)、小圆细胞及中等大小多边形或圆形细胞,区域可见典型的横纹肌样细胞(图4),部分围绕血管排列,部分富有黏液样背景,有不同程度坏死(图5~7)。肿瘤细胞边界清楚,核圆形或梭形,染色深,可见核仁,部分偏位,胞质红染,呈横纹肌样,病理性核分裂象易见。
2.3 免疫表型肿瘤细胞Olig2(5/10)、p53(5/10)、Syn(5/10)、GFAP(4/10)、EMA(3/10)、CD99(3/10)和vimentin(3/10)均阳性,10例INI1(图8)、H3K27M和CD34均阴性,Ki-67增殖指数为20%~60%(图9)。
3 讨论
3.1 诊断AT/RT是一种少见、预后差的CNS胚胎性肿瘤,具有高度侵袭性,常见于婴幼儿、儿童,大部分患者小于3岁,6岁以上及成人患者少见[1-2]。本组10例患者,年龄1~6岁,平均2.5岁,男女童各5例。AT/RT在幕上常位于大脑半球的额顶叶,其次是脑室、鞍上及松果体。幕下常好发于小脑、桥小脑角及脑干,临床症状与发病部位、年龄相关,有文献报道肿瘤位于小脑者表现为行走不稳,位于脑室者以呕吐为主要症状,位于大脑半球者表现为头痛[3]。本组10例,2例发生于小脑,表现为行走不稳、肢体麻木等症状;5例发生于脑室,也以呕吐、行走不稳为主要症状;3例发生于额顶叶,有头晕、呕吐的症状,1例伴视力模糊,1例伴抽搐症状。AT/RT影像学表现为占位性病变,可伴囊性变、出血,增强扫描后均有不规则或不均匀强化,也可有不同程度的脑积水改变,但非该病特有的影像学表现,不易与其他CNS的肿瘤鉴别。
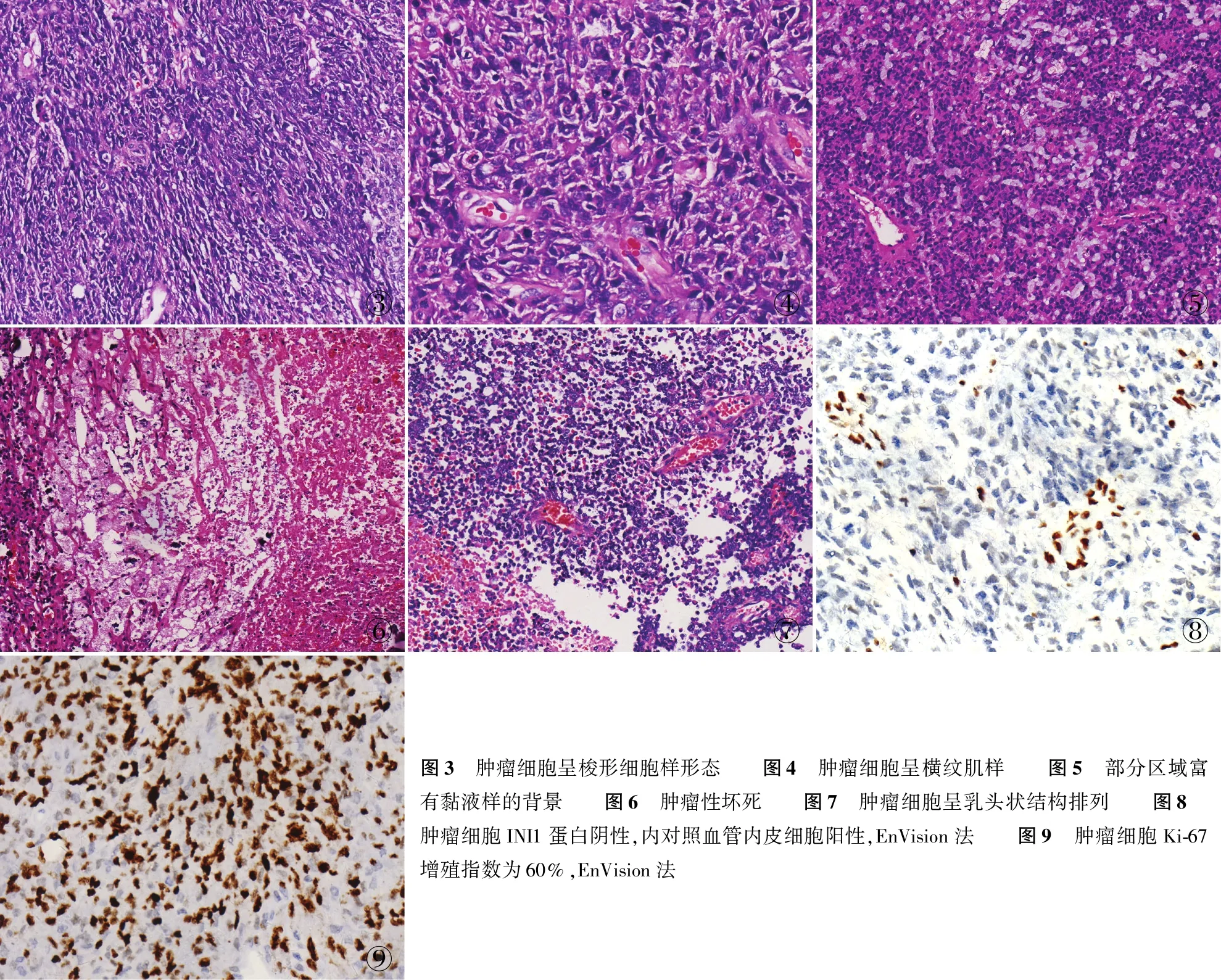
③④⑤⑥⑦⑧⑨图3 肿瘤细胞呈梭形细胞样形态 图4 肿瘤细胞呈横纹肌样 图5 部分区域富有黏液样的背景 图6 肿瘤性坏死 图7 肿瘤细胞呈乳头状结构排列 图8 肿瘤细胞INI1蛋白阴性,内对照血管内皮细胞阳性,EnVision法 图9 肿瘤细胞Ki-67增殖指数为60%,EnVision法
目前研究显示,AT/RT起源于多潜能胚胎细胞,肿瘤细胞可表达神经、上皮及间叶标记;也有学者提出其可能是起源于神经节神经胶质或者一些低级别中枢神经病变,但仍需进一步研究证实。镜下见肿瘤组织形态较为复杂,具有多向分化的特点,可见典型横纹肌样细胞及不同程度坏死,可见梭形细胞呈假菊形团形成,部分围绕血管排列[1]。10例AT/RT肿瘤细胞弥漫成片,部分围绕血管排列,部分富有黏液样背景,有不同程度的坏死。肿瘤细胞呈多形性,有梭形细胞、小圆细胞及中等大小多边形或圆形细胞,区域可见典型的横纹肌样细胞,肿瘤细胞边界清楚,核圆形或梭形,染色深,可见核仁,部分偏位,胞质红染,呈横纹肌样,病理性核分裂象易见,大部分AT/RT病理性核分裂象易见,Ki-67增殖指数较高,本组10例高达20%~60%。
肿瘤细胞具有多种免疫表型特性,GFAP、EMA、Syn、vimentin、CD99等可呈阳性,提示该肿瘤呈多向分化。研究证实AT/RT有特征性的遗传学改变,即为编码INI1蛋白的SMARCB1(又称hSNF5/INI1)基因或编码BRG1蛋白的SMARCA4(又称BRG1)基因发生缺失,由于这两个基因是哺乳动物SWL/SNF复合物的组成成分,通过ATP依赖的方式改变染色体结构,从而改变基因表达,促进肿瘤形成[4-5]。因INI1蛋白或BRG1蛋白的表达缺失与INI1基因或BRG1基因的缺失状态基本一致,所以INI1蛋白或BRG1蛋白免疫组化检测可作为诊断AT/RT的金标准[6]。本组10例INI1蛋白表达均缺失,支持AT/RT的诊断。
3.2 鉴别诊断
3.2.1其他CNS胚胎性肿瘤 (1)髓母细胞瘤:经典型者组织形态相对单一,大细胞型与AT/RT类似,但无肿瘤性上皮样和梭形细胞成分,缺乏明显核仁,区域可伴有神经元或胶质分化,免疫表型也不及AT/RT的多样化,无INI1或BRG1基因失活突变。髓母细胞瘤常伴脑脊液播散和CNS外转移。(2)有多层菊形团的胚胎性肿瘤(embryonal tumor with multilayered rosettes, ETMR)[1]:组织学具有特征性的多层菊形团结构及各种变异形,肿瘤大部分区域为原始胚胎成分,免疫组化标记vimentin、NES、EMA阳性及CD99灶阳性,提示有不同程度的分化,具有神经毡的肿瘤细胞Syn和NeuN呈强阳性,但标记菊形团的细胞神经元和胶质细胞均阴性,所有ETMR的INI1核强阳性及LIN28A强阳性,根据有无C19MC基因改变分为:ETMR,C19MC变异型和ETMR,非特指。此外,ETMR可沿软脑膜广泛播散,也可向颅外软组织侵袭性生长,甚至向颅外远处转移。(3)髓上皮瘤:类似于胚胎性神经管的恶性肿瘤,特征性病变为假复层上皮细胞呈乳头状、管状排列,似原始神经管结构。神经上皮也可向不同方向分化,肿瘤细胞vimentin、EMA阳性,但神经上皮肿瘤细胞GFAP、NSE呈阴性。而与神经上皮不同区域的肿瘤细胞可向神经元、星形细胞、少突胶质细胞或室管膜母细胞分化,免疫表型可以表达相应胚系的标记,也无INI1或BRG1基因失活突变。髓上皮瘤可伴有脑脊液播散,但很少发生全身转移。(4)有横纹肌样特征的CNS胚胎性肿瘤:其好发年龄和部位、组织学形态及生物学行为与AT/RT相同,但无INI1或BRG1基因失活突变[1,7]。
3.2.2上皮样胶质母细胞瘤 常见于儿童及青年人,位于各个脑叶,以颞叶、额叶及间脑常见,呈侵袭性生长,常早期复发,易出现瘤内出血及脑脊液播散。肿瘤起源尚不清楚,以实性病变为主,与周边脑组织界限清晰。镜下主要为宽胞质的上皮样细胞,成片密集排列的上皮样肿瘤细胞间无神经毡样结构,可以有横纹肌样细胞构成,伴有显著的核分裂象、血管内皮细胞增生及坏死,多为凝固性坏死。周边相邻组织可见低级别胶质瘤成分。肿瘤细胞vimentin、S-100和INI1均阳性,GFAP、Olig2可阳性或阴性,Syn、NF可局灶阳性,上皮标记表达不恒定,但不表达间叶标记。约50%病例伴BRAF V600E基因突变,Ki-67增殖指数较高,IDH呈野生型及无ATRX基因突变[1,8]。
3.2.3横纹肌肉瘤 CNS横纹肌肉瘤均为胚胎型,组织学特点为具有胚胎样未分化的间叶细胞、不同分化阶段的横纹肌母细胞及黏液样变基质,这些肿瘤细胞desmin、Myogenin及MyoD1均阳性,但缺乏上皮和神经的标记有助于鉴别[9]。
3.2.4横纹肌样脑膜瘤 横纹肌样脑膜瘤仅见于脑膜瘤复发病例,影像学上脑膜瘤有特征性的“脑膜拖尾征”,免疫组化标记GFAP、SMA、Syn均阴性,INI1核阳性,Ki-67增殖指数较低,即使间变性脑膜瘤也低于25%,而AT/RT的Ki-67增殖指数往往较高,甚至高达80%[1,10]。
3.2.5恶性黑色素瘤 恶性黑色素瘤的形态复杂多样,可具有丰富的胞质,核大及显著的核仁,核偏位类似于横纹肌样细胞,但部分恶性黑色素瘤伴有黑色素,免疫组化标记HMB-45及Melan-A阳性,GFAP、CK和EMA均阴性,可与CNS的AT/RT鉴别[11]。
3.3 预后及随访AT/RT是一种好发于婴幼儿的高度恶性的CNS胚胎性肿瘤,病程进展迅速、预后差,本组10例患者中位生存时间小于6个月。患者年龄小于3岁,病变有播散及转移提示预后差,约一半的患者可出现脑脊液播散。目前尚无标准化的治疗方案,治疗以手术最大限度切除为主,术后辅以放、化疗,随着多模式治疗方法应用于临床,如骨髓移植、干细胞支持、免疫治疗PDL-1的推广,效果均不佳,AT/RT的5年无进展生存期和总生存期并无改善[12]。
