Microbial diversity assembled from series-diluted suspensions of diseasesuppressive soil determines pathogen invasion resistance
2021-03-17YannanOU,ZongzhuanSHEN,BeibeiWANG等
Dear Editor,
Soil microbial biodiversity loss caused by agricultural intensification,climate change,and the application of chemical fertilizer has become a serious problem that threatens humans(Wallet al.,2015).One phenomenon responsible for economic and food security issues is soil-borne diseases(Fisheret al.,2012),which were reported to be associated with microbial diversity loss(Shenet al.,2013;Fuet al.,2017).Suppressive soils provide an ideal model for studying the opposing effect of the microbial community against soil-borne pathogen invasion(Schlatteret al.,2017).Our previous work has demonstrated that a suppressive soil shows higher microbial richness than a conducive soil(Shenet al.,2015).Hence,a better understanding of whether microbial diversity is truly a mechanism contributing to the inhibition of pathogen establishment in suppressive soils could contribute to pathogen management.In addition,mixed populations can perform complex functions and are robust to changes in the environment,and synthetic communities retain key features of natural ones(Großkopf and Soyer,2014),giving us an opportunity to manipulate microbiome assembly at the initial stages.Thus,beyond extremely high diversity,the identification of species or groups of microbes that are responsible for suppression will help to obtain a more comprehensive picture.
Based on the diversity-invasion theory,we hypothesized that microbial diversity plays an important role in disease suppression through certain key taxa.To test this hypothesis,we used a dilution method to reestablish the microbial community in two sterilized soil sources(disease-conducive and-suppressive soils)viainoculation of disease-suppressive soil microbes.Then we introduced the same abundance ofFusarium oxysporumf.sp.cubense(Foc)to each sample(see Supplementary Material for details of the experimental procedure)to test the suppressive ability.
At the end of the assembly phase(Fig.1a)(two months after microbe inoculation),no significant difference was detected in bacterial abundance within reestablished conducive soils(P=0.085)or suppressive soils(P=0.374)(Fig.1b).However,bacterial abundance(Fig.1b)and richness(Fig.1b)were significantly lower in the reestablished conducive soils than in the suppressive soils(P<0.05).Additionally,the soil source explained 79% of the total variance of soil community composition(Fig.1d,Table SI,see Supplementary Material for Table SI).That is,during bacterial community establishment,the physicochemical environment was most important,followed by the initial microbial seed.Research has also shown that soil physicochemical properties are the main factors affecting microbe establishment(Xunet al.,2015).Thus,abiotic conditions appropriate for suppressive microbes should be the first thing to consider when selecting and designing suppressive microbes for agroecosystems ensuring their functions.
In the reestablished suppressive soils,bacterial richness was significantly lower in the high-level dilution(d3)treatment than in the other dilution treatments(P<0.05)(Fig.1b).Additionally,the assessment ofFocinhibition ability showed thatFocabundance was significantly lower in the low-level dilution(d1)and moderate-level dilution(d2)treatments than in the d3 treatment(P<0.05)(Fig.2a).Although soil physicochemical properties and microbial diversity may both influence the survival of introduced strains in reestablished conducive soils,results from our reestablished suppressive soils highlighted that after incubation,bacterial richness decreased and species abundance changed,resulting in loss ofFocinhibition ability.Furthermore,a significant negative relationship was observed between bacterial richness andFocabundance(r=−0.733,P<0.05)(Fig.2b).In accordance with our result,Huet al.(2016)showed increased disease suppression with increased species richness in assembled communities containing eight differentPseudomonasstrains.Synthetic microbial communities reported by van Elsaset al.(2012)also indicate that soil bacterial diversity controls bacterial pathogen establishment.
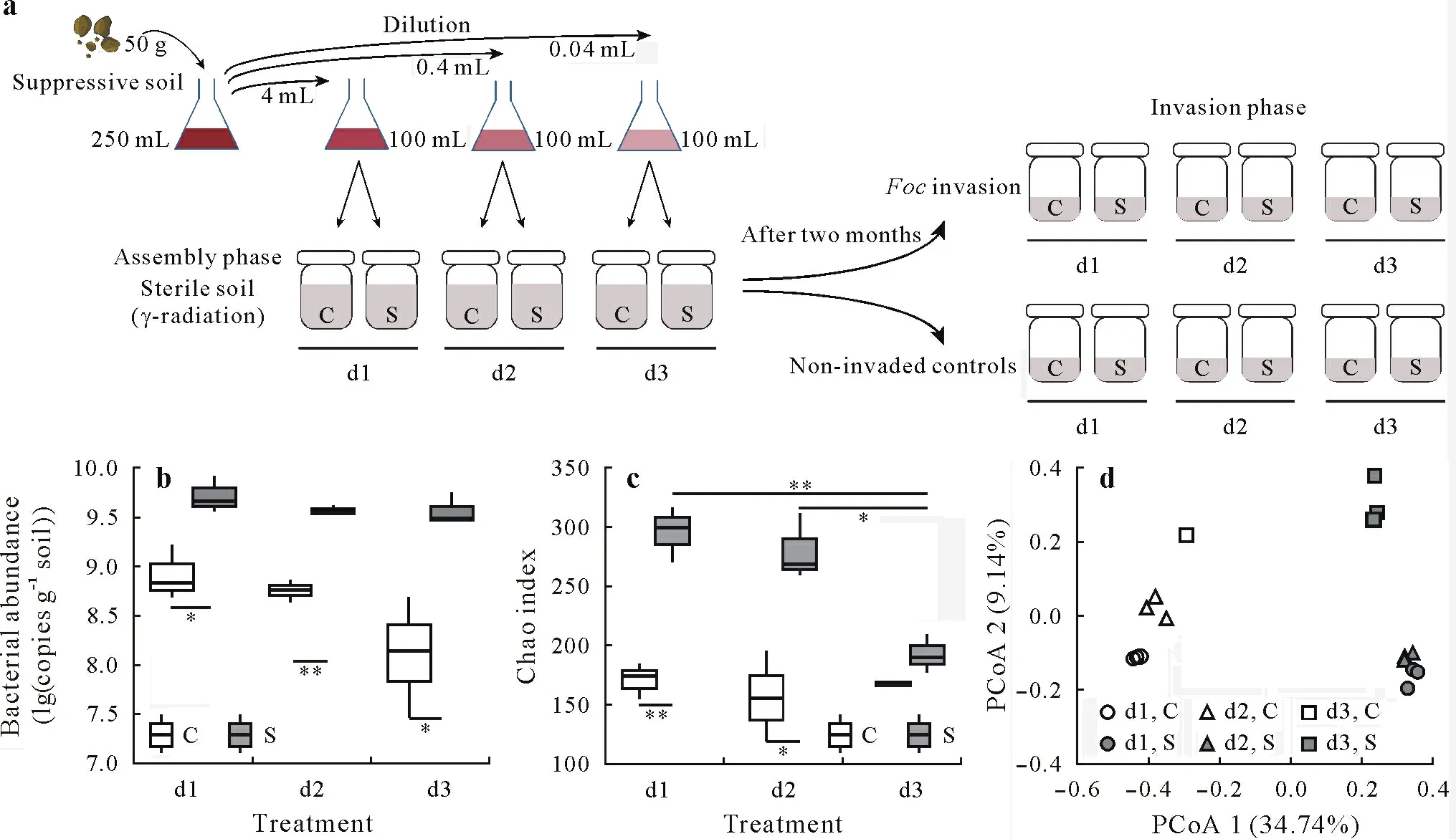
Fig.1 Design of the experiment composed of different dilution treatments,low-level dilution(d1),moderate-level dilution(d2),and high-level dilution(d3)treatments,with or without Fusarium oxysporum f.sp.cubense(Foc)invasion two months after microbe inoculation and the soil bacterial community at the end of the assembly phase:the experimental setup,in which each bottle setup was in triplicate(a);the bacterial abundance(b)and richness(Chao index)(c)based on estimated absolute abundance two months after microbe inoculation;and the bacterial community composition calculated from the Bray-Curtis distance and shown in principal coordinate analysis(PCoA)ordinations based on estimated absolute abundances two months after microbe inoculation(d).Two d3 samples were excluded after two months of assembly in conducive soil due to a very high(87.5%)relative abundance of one amplicon sequence variant(ASV)in one sample(Fig.S1,see Supplementary Material for Fig.S1)and a very large number of ASVs in the other sample(Fig.S2,see Supplementary Material for Fig.S2).The preincubated suppressive soil(50 g)was suspended in 250 mL of sterile water,which was treated as an undiluted suspension(d0 treatment),and then 4(d1),0.4(d2),or 0.04(d3)mL suspension of the d0 treatment was added into 100 mL sterilized water.C represents the disease-conducive soil,S represents the disease-suppressive soil,and the asterisks*and**indicate significant differences at P<0.05 and P<0.01,respectively(t-test).
Furthermore,random forest classification was performed to correlate theFocinhibition ability with the bacterial richness and the estimated absolute abundance of bacterial taxa at the family level in reestablished suppressive soils.The random forest classification method is a robust machine-learning algorithm for classification and regression(Breiman,2001).The model showed 77.8% accuracy.Among the 24 biomarkers,7 richness biomarkers and 3 abundance biomarkers were significantly higher in the d1 and d2 treatments than in the d3 treatment in reestablished suppressive soils(P<0.05)(Fig.S3,see Supplementary Material for Fig.S3).Furthermore,the richness of Chitinophagaceae(r=−0.687),Cytophagaceae(r=−0.646),Gemmatimonadaceae(r=−0.791),Myxococcaceae(r=−0.728),Oxalobacteraceae(r=−0.600),and Planctomycetaceae(r=−0.707)and the abundance of Opitutaceae(r=−0.625)and Sphingobacteriaceae(r=−0.570)were significantly negatively correlated toFocabundance in all analyzed soil samples(P<0.05)(Fig.S4,see Supplementary Material for Fig.S4).In detail,Chitinophaga,Flavitalea,andTerrimonasin Chitinophagaceae,Nibribacterin Cytophagaceae,Gemmatimonasin Gemmatimonadaceae,Aggregicoccus,Angiococcus,andCorallococcusin Myxococcaceae,Oxalicibacteriumin Oxalobacteraceae,andSingulisphaeraandBlastopirellulain Planctomycetaceae may be important because they were only found in the d1 and d2 treatments of reestablished suppressive soils(Fig.2d,e).Also,culturable strains were reported in these genera(Zhanget al.,2003;Schlesneret al.,2004;Sahinet al.,2010;Kulichevskayaet al.,2012;Kanget al.,2013;Garcia and Müller,2014;Rosenberg,2014;Soodet al.,2015;Kim S Jet al.,2016;Kim Y Jet al.,2016).
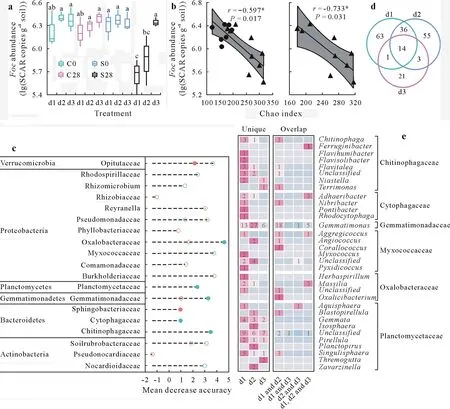
Fig.2 Associations of microbial groups with Fusarium oxysporum f.sp.cubense(Foc)abundance:the abundance of Foc in different dilution(d1-d3)treatments(a),relationships between bacterial richness(Chao index)and Foc abundance(b),results of random forest classification showing the relative contribution of the bacterial richness or estimated absolute abundance(EAA)of families(two months after microbe inoculation)in determining the abundance of Foc(28 d after Foc inoculation)(c),overlap of amplicon sequence variants(ASVs)from 6 families whose richness is important for resistance to Foc abundance from c(d)and heat map showing the number of ASVs from d and their genus information.In a,different letters on the labels indicate significant(P<0.05)differences between samples based on one-way analysis of variance(ANOVA)and a post-hoc paired t-test with false discovery rate(FDR)correction(n=3),C0 and S0 represent Day 0 after Foc inoculation of disease-conducive soil and disease-suppressive soil,respectively,C28 and S28 represent Day 28 after Foc inoculation of disease-conducive soil and disease-suppressive soil,respectively,and SCAR indicates sequence-characterized amplified region.In b,the circle represents conducive soil,the triangle represents suppressive soil,and the asterisk*denotes a significant correlation at P<0.05.In c,the mean decrease accuracy indicates the prediction accuracy of each taxon(P<0.001),the red and green circles represent the EAA and the richness biomarkers,respectively,and the solid circles represent that a biomarker is significantly higher in the d1 and d2 treatments than in the d3 treatments in suppressive soil(Fig.S3,see Supplementary Material for Fig.S3)and a significant negative relationship to Foc abundance(Fig.S4,see Supplementary Material for Fig.S4).In e,detailed numbers of ASVs were listed in heat map,and a blank indicates a value of zero.See Fig.1 for details of the d1-d3 treatments.
For eight predictors from the model,their importance in ecosystem functioning(resistance toFocabundance)was highlighted.Oxalobacteraceae are more abundant in the sugar beet rhizosphere uponRhizoctonia solaniinvasion(Chapelleet al.,2016).Species belonging to the families Oxalobacteraceae(Adrangiet al.,2010),Cytophagaceae(Jounget al.,2014),Chitinophagaceae(Rosenberg,2014),and Sphingobacteriaceae(Matsudaet al.,2001;Songet al.,2017)have been reported to degrade chitin,which is a component of theFusariumcell wall.Additionally,a long-term(8 years)field experiment showed suppression ofVerticillium dahliaeupon the addition of chitin,which may be due to the higher abundance of Actinobacteria and Oxalobacteraceae and the expression of the chitinase genechiA(Cretoiuet al.,2013).Many species within Myxococcaceae are referred to as myxobacteria;the compounds that they produce can disrupt fungal macromolecule synthesis(Knauth and Reichenbach,2000;Bodeet al.,2003;Kunzeet al.,2008),and the exoenzymes that they produce allow them to use a range of biological macromolecules and microorganisms(Reichenbach,2001;Kaiser,2003).Although Opitutaceae andGemmatimonasfrom Gemmatimonadaceae have been shown to present higher abundance in healthy soils(Liet al.,2015;Wuet al.,2015),no biological association of these groups or the family Planctomycetaceae withFocinhibition has been reported thus far.Many studies have revealed that antagonistic interactions help plants in pathogen defense(Mendeset al.,2011;Chaet al.,2016).Thus,the results in this research might be useful in expanding our knowledge to establish a functional synthetic community to inhibit pathogen invasion.Overall,our study revealed that microbial diversity indeed affects pathogen inhibition ability.The richness of Chitinophagaceae,Cytophagaceae,Myxococcaceae,and Oxalobacteraceae and the abundance of Sphingobacteriaceae were negative biomarkers ofFocabundance showing ecological importance.Therefore,these findings should guide future efforts linking environmental conditions with microbiome establishment and the choice of species for artificial community assembly.
ACKNOWLEDGEMENT
This research was supported by the National Natural Science Foundation of China(Nos.31572212,31601836,and 31672239),the Fundamental Research Funds for the Central Universities of China(Nos.KJQN201746 and KYT201802),the Priority Academic Program Development of Jiangsu Higher Education Institutions(PAPD),China,the 111 Project of China(No.B12009),the Innovative Research Team Development Plan of the Ministry of Education of China(No.IRT_17R56),and the China Postdoctoral Science Foundation(Nos.2016M590469 and 2018T110509).
SUPPLEMENTARY MATERIAL
Supplementary material can be found in the online version.
杂志排行

Pedosphere的其它文章
- Notes to Authors
- Agricultural and environmental challenges in agroecosystems
- In Commemoration of Professor Tianren YU’s 100th Anniversary
- Global patterns of phosphorus transformation in relation to latitude,temperature and precipitation
- Chemical and spectroscopic characteristics of humic acid from a clay loam soil in Ontario after 52 years of consistent fertilization and crop rotation
- Interactive effects of elevated CO2 and nitrogen fertilization levels on photosynthesized carbon allocation in a temperate spring wheat and soil system