赤胫散指纹图谱的建立及其3种化学成分含量的测定*
2021-03-13王彬宇黄家宇杜秋莹陈思忆鲁婷婷李莉
王彬宇, 黄家宇, 杜秋莹, 陈思忆, 鲁婷婷, 李莉*
(贵州医科大学 药学院, 贵州 贵阳 550025)
赤胫散又名花蝴蝶、散血草,为蓼科蓼属植物赤胫散(PolygonumruncinatumBuch.-Ham.var.sinenseHemsl.)的干燥根茎,是西南少数民族地区民间常用药[1-2],其性寒,味苦、涩,用于治疗跌扑损伤、痈疖肿毒及风湿痹痛等,具有清热解毒、活血舒筋等功效[3-5]。赤胫散现收载于《贵州省中药材、民族药材质量标准》2003年版中,其鉴别方法为与对照药材薄层色谱法(thin layer chromatography ,TLC)定性鉴别,无含量测定项[6]。目前,国内外对赤胫散的质量研究较少,难以全面评价药材质量[7-9]。赤胫散主要含有酚酸、挥发油类成分[10-14],其中没食子酸具有抗炎抗氧化的药理作用[15];鞣花酸和3,3′-二甲基鞣花酸具有抗菌抗病毒活性[16-18],毒副作用小,与赤胫散功能主治一致。基于此,本实验通过建立赤胫散高效液相色谱法(high performance liquid chromatography,HPLC)指纹图谱,并结合化学计量学手段[19-26]进行聚类分析、主成分分析及正交偏最小二乘判别分析,同时对没食子酸、鞣花酸及3,3′-二甲基鞣花酸的含量进行测定,为其质量控制提供实验依据。
1 材料与方法
1.1 材料
1.1.1药材来源 赤胫散药材17批均为本省野生(表1),经贵州医科大学药学院生药学教研室龙庆德副教授鉴定为蓼科蓼属植物赤胫散的干燥根茎。
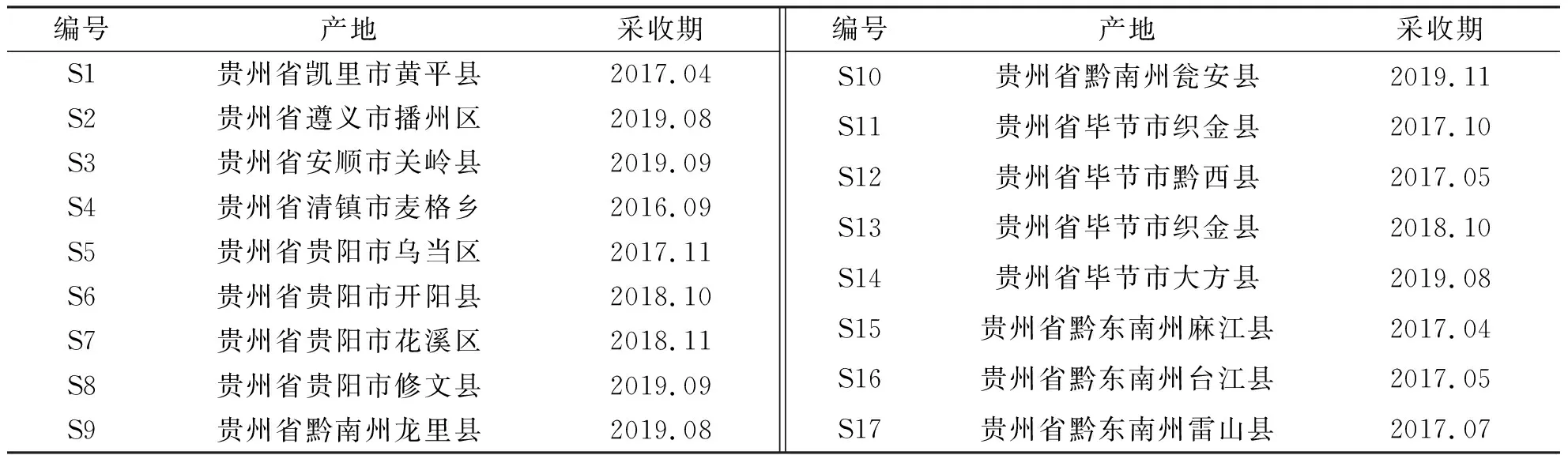
表1 17批赤胫散样品来源信息Tab.1 Source information of 17 batches of P. runcinatum
1.1.2主要试剂与仪器 没食子酸对照品(批号110831-201204,中国食品药品检定研究院),鞣花酸对照品(批号GZDD-0272,纯度≥98%,贵州迪大科技有限责任公司),3,3′-二甲基鞣花酸(批号160810,北京北纳创联生物技术研究院),水为纯净水,磷酸为分析纯,乙腈为色谱纯,其余试剂均为分析纯;ULtiMate 3000 型高效液相色谱仪[赛默飞世尔科技(中国)有限公司],MS105DU型电子分析天平(瑞士Mettler-Toledo公司),KQ3200E型超声波清洗器(昆山市超声仪器有限公司)。
1.2 方法
1.2.1色谱条件 色谱柱为ACE C18-AR(250 mm×4.6 mm,5 μm),检测波长为254 nm,流速为1 mL/min,柱温为30 ℃,进样量为10 μL;流动相为乙腈(A)-0.1%磷酸溶液(B);梯度洗脱为0~6 min、5%~15%A,6~20 min、15%~17%A,20~45 min、17%~30%A,45~60 min、30%~70%A。
1.2.2对照品溶液的制备 分别取没食子酸、鞣花酸及3,3′-二甲基鞣花酸对照品适量,精密称定,加甲醇制成浓度为1.12、1.11及0.96 g/L的单一对照品储备液;分别精密量取上述对照品储备液适量,加甲醇稀释制成混合对照品溶液(每升含没食子酸39.20 mg、鞣花酸255.30 mg及3,3′-二甲基鞣花酸28.80 mg)。
1.2.3供试品溶液的制备 取赤胫散药材粗粉(过60目筛)约0.3 g,精密称定,置100 mL具塞锥形瓶中,精密加70%甲醇25 mL,密塞,称定质量,37 ℃超声(功率150 W,频率40 kHz)处理45 min,放冷,70%甲醇补足减失质量,经0.22 μm有机微孔滤膜过滤,取续滤液,即得供试品溶液。
1.2.4指纹图谱的建立 (1)精密度试验:取赤胫散药材粉末(编号S9),按“1.2.3”项下方法制备供试品溶液,按“1.2.1”项下色谱条件连续进样测定6次,以鞣花酸的保留时间和峰面积为参考,计算各共有峰相对标准偏差(rlative sandard deviation ,RSD)值。(2)重复性试验:取赤胫散药材粉末(编号S9),按“1.2.3”项下方法平行制备供试品溶液6份,按“1.2.1”项下色谱条件进行进样测定,以鞣花酸的保留时间和峰面积为参考,计算各共有峰RSD值。(3)稳定性试验:取赤胫散药材粉末(编号S9),按“1.2.3”项下方法制备供试品溶液,室温下放置,于0、2、4、8、12及24 h时按“1.2.1”项下色谱条件进行进样测定,以鞣花酸的保留时间和峰面积为参考,计算各共有峰RSD值。(4)赤胫散指纹图谱的建立:分别取17批赤胫散药材粉末,按“1.2.3”项下方法制备供试品溶液,按“1.2.1”项下色谱条件进行进样测定,采用中药色谱指纹图谱相似度评价系统(2012年版)软件,将S1样品色谱图设为参照图谱,中位数法,时间窗宽度0.2 min,生成对照图谱,进行指纹图谱的建立、共有峰的指认与相似度分析。
1.2.5含量测定 (1)线性关系考察:取“1.2.2”项下混合对照品溶液适量,加甲醇逐级稀释,制成系列浓度混合对照品溶液,按“1.2.1”项下色谱条件进样测定,以各成分的质量浓度为横坐标,峰面积为纵坐标,绘制线性标准曲线,得到3种成分各自线性范围及相关系数(r)。(2)精密度试验:取赤胫散药材粉末(编号S9),按“1.2.3”项下方法制备供试品溶液,以“1.2.1”项下色谱条件进样测定6次,记录峰面积,计算3种成分RSD值。(3)重复性试验:取赤胫散药材粉末(编号S9),按“1.2.3”项下方法平行制备供试品溶液6份,以“1.2.1”项下色谱条件进样测定,记录峰面积,计算3种成分RSD值。(4)稳定性试验:取赤胫散药材粉末(编号S9),按“1.2.3”项下方法制备供试品溶液,室温下放置,于0、2、4、8、12、24 h时以“1.2.1”项下色谱条件进行进样测定,记录峰面积,计算3种成分RSD值。(5)加样回收率试验:精密称取已知含量的同一批赤胫散药材粉末(编号S9)0.15 g,共6份,分别置于100 mL具塞锥形瓶瓶中,加入各单一对照储备液适量,按“1.2.3”项下方法制备供试品溶液,按“1.2.1”项下色谱条件进行进样测定,记录峰面积,并计算3种成分的加样回收率及其RSD值。
1.3 统计学分析
应用SPSS 23.0软件,采用组间联接法、选择欧式平方距离为度量,对17批赤胫散共有峰峰面积数据进行聚类分析;应用SIMCA 14.1软件对17批赤胫散样品共有峰峰面积进行主成分分析,绘制主成分分析得分图(以特征值>1作为标准,计算其累积方差贡献率与特征值),同时进行正交偏最小二乘判别分析,得到得分图[以变量重要性投影值(very important person,VIP)值>1作为标准,得到可能引起赤胫散样品间质量差异的主要标志性成分]。
2 结果
2.1 赤胫散药材指纹图谱的建立和方法学考察
2.1.1指纹图谱方法学 精密度试验中,测得各共有峰的相对保留时间RSD<0.09%,相对峰面积RSD<2.58%,说明仪器精密度良好。重复性试验中,测得各共有峰的相对保留时间RSD<0.18%,相对峰面积RSD<2.97%,说明方法重复性良好。稳定性试验中,测得各共有峰的相对保留时间RSD<0.23%,相对峰面积RSD<2.92%,表明样品在室温放置24 h内稳定性良好。
2.1.2赤胫散药材指纹图谱的建立和共有峰的指认 经多点校正后进行色谱峰重叠匹配,标定了19个共有峰(图1)。将样品对照指纹图谱与对照品的保留时间和紫外吸收光谱比对,指认了3种成分,分别为2号峰(没食子酸)、11号峰(鞣花酸)、19号峰(3,3′-二甲基鞣花酸),其中11号峰(鞣花酸)为所有样品中均含有,分离度较好,保留时间适中且峰面积最大,因此选其作为参照峰(S,图2)。对17批赤胫散样品进行相似度计算,结果17批样品指纹图谱与对照指纹图谱的相似度为0.768~0.997(表2),表明各产地样品间整体质量存在一定的差异。

注:1~19为共有峰编号。图1 赤胫散样品(17批)HPLC指纹图谱叠加图(S1~S17)及对照图谱(R)Fig.1 HPLC fingerprints(S1~S17)and control fingerprint(R)of 17 batches of P. runcinatum

注:1~19为共有峰编号。图2 对照品和对照指纹图谱色谱图Fig.2 HPLC chromatogram of substance control and control fingerprint

表2 赤胫散样品(17批)相似度评价结果Tab.2 Results of similarity evaluation of 17 batches of P. runcinatum
2.2 化学模式识别分析
2.2.1聚类分析 当距离为15时,可将17批样品聚为2类,其中Ⅰ类包括S1~S6、S8、S9、S15及S17,Ⅱ类包括S7、S10~S14及S16。见图3。

图3 赤胫散样品(17批)聚类分析图Fig.3 Hierarchical cluster analysis for 17 batches of P. runcinatum
2.2.2主成分分析 主成分分析结果显示,以特征值>1作为标准时,得到5个主成分,其累积方差贡献率为89.427%;可将17批赤胫散样品分为2类,其结果与聚类分析一致,S1~S6、S8、S9、S15及S17大多集中于一、四象限,S7、S10~S14及S16集中于二、三象限。见表3和图4。
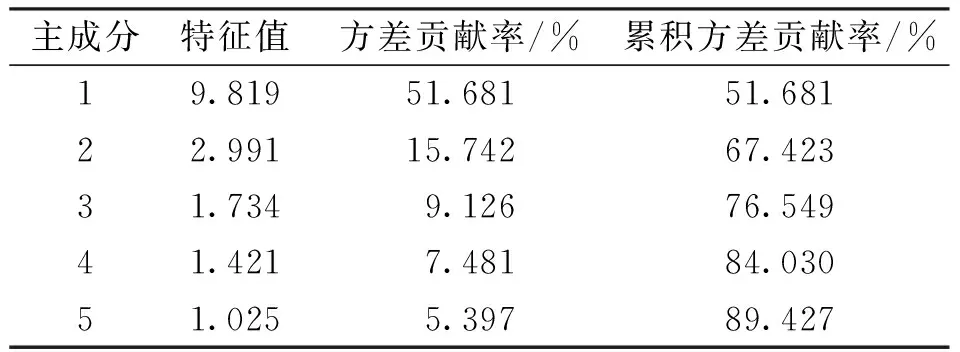
表3 主成分分析方差贡献率与特征值Tab.3 Variance contribution rate and eigenvalue and of principal component analysis

注:t[1]为主成分1,t[2]为主成分2。图4 赤胫散样品(17批)主成分分析Fig.4 Score scatter plot of principal component analysis of 17 batches of P. runcinatum
2.2.3正交偏最小二乘判别分析 正交偏最小二乘判别分析结果表明,17批样品可分为2类,结果与聚类分析、主成分分析一致;模型参数R2X(cum)=0.832,R2Y(cum)=0.980,均>0.5,说明模型稳定且预测性好;绘制VIP图,以VIP>1作为标准,色谱峰9、8、14、4、5、11、7、13、12及10为引起赤胫散样品间质量差异的主要标志性成分。见图5和图6。
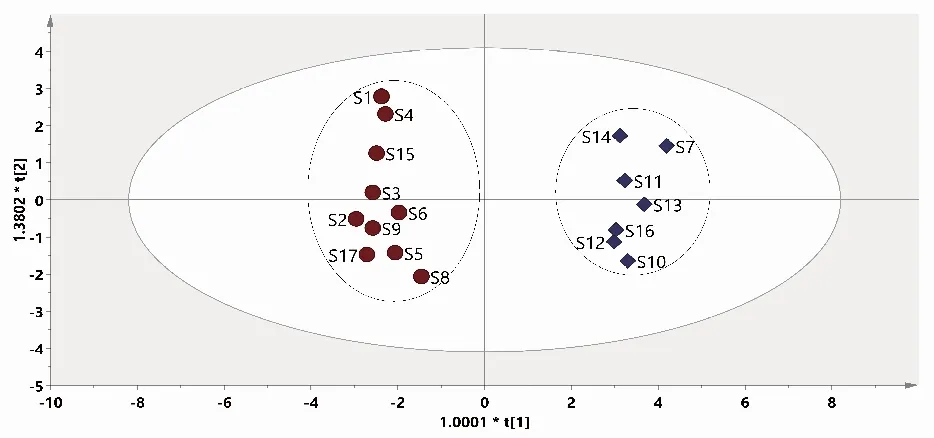
注:t[1]为主成分1,t[2]为主成分2。图5 赤胫散样品(17批)正交偏最小二乘判别分析Fig.5 Score scatter plots of orthogonal partial least square-discriminant analysis of 17 batches of P. runcinatum
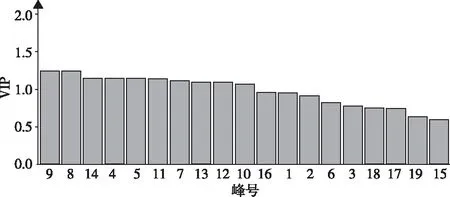
图6 赤胫散样品(17批)正交偏最小二乘判别分析VIP值Fig.6 VIP values of common peaks in Orthogonal partial least square-discriminant analysis model of 17 batches of P. runcinatum
2.3 3种成分含量测定与方法学考察
2.3.1含量测定方法学考察结果 线性关系考察中,没食子酸、鞣花酸及3,3′-二甲基鞣花酸在各自范围内线性关系良好(表4);精密度试验中,测得没食子酸、鞣花酸及3,3′-二甲基鞣花酸峰面积的RSD分别为0.38%、0.05%及2.01%,说明仪器精密度良好;重复性试验中,测得没食子酸、鞣花酸及3,3′-二甲基鞣花酸峰面积的RSD分别为1.06%、0.18%及0.32%,说明方法重复性良好;稳定性试验中,测得没食子酸、鞣花酸及3,3′-二甲基鞣花酸峰面积的RSD分别为1.80%、0.56%及0.17%,表明样品在室温放置24 h内稳定性良好;加样回收率试验中,平均回收率分别为103.48%、96.90%及98.61%,RSD值均<2.21%,说明加样回收率良好(表5)。

表4 各成分线性范围与回归方程Tab.4 Regression equation and linear range of 3 components
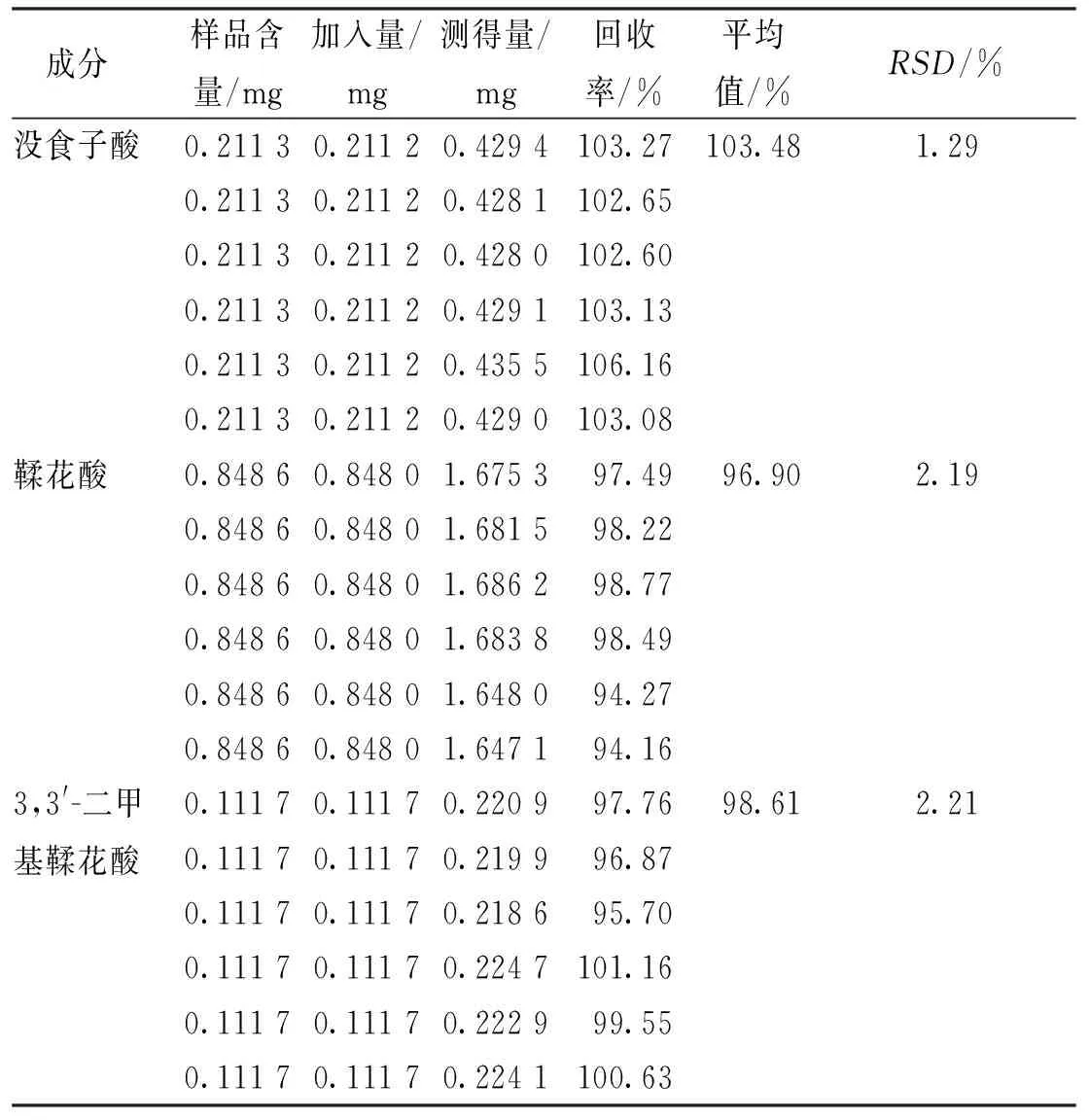
表5 各成分加样回收率试验结果Tab.5 Results of recovery test of 3 components
2.3.23种成分含量测定结果 含量测定结果表明,不同产地赤胫散中3种成分含量差异较大,见表6。
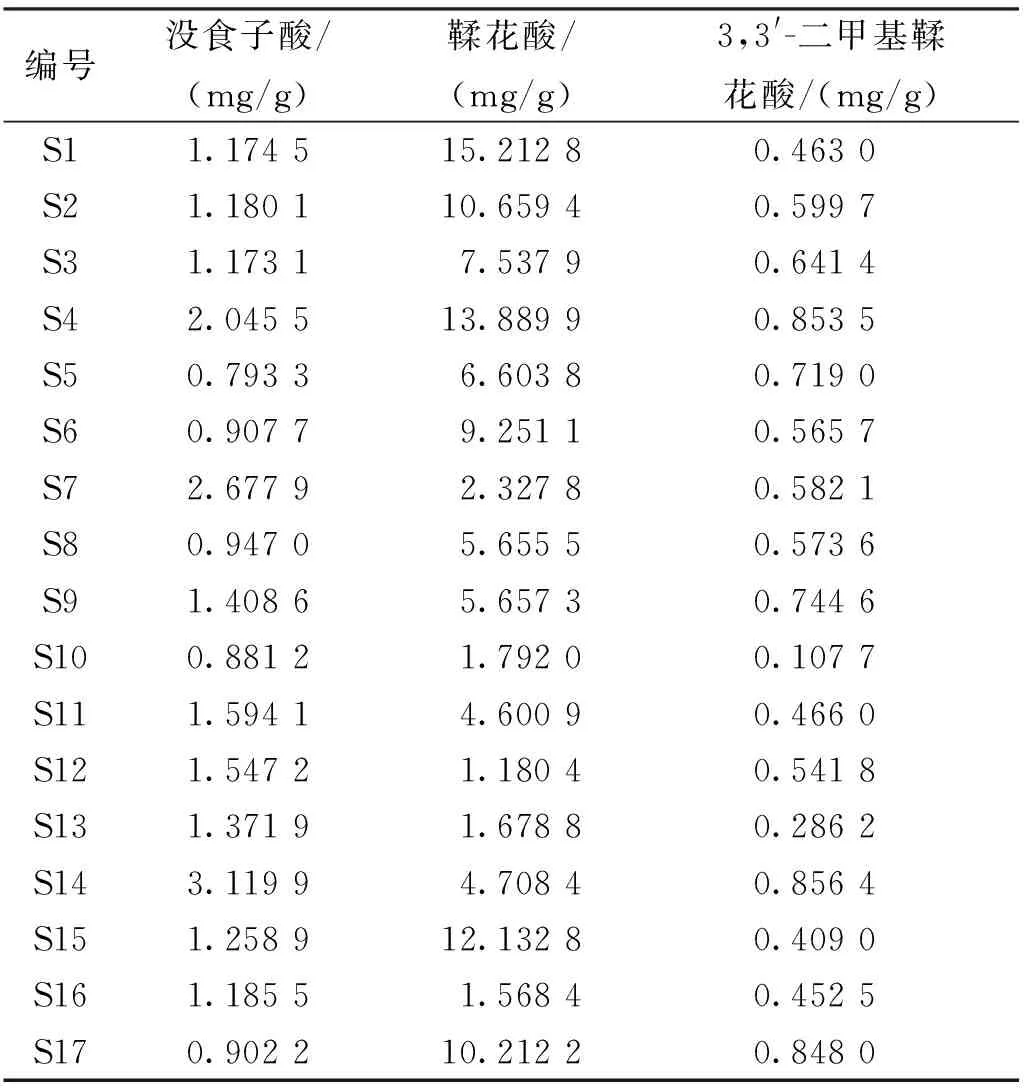
表6 赤胫散样品(17批)3种成分含量测定结果Tab.6 Results of content determination of 3 components of 17 batches of samples
3 讨论
本实验前期分别考察了8种提取溶剂(30%乙醇、50%乙醇、70%乙醇、95%乙醇、30%甲醇、50%甲醇、70%甲醇及100%甲醇),其中70%甲醇提取的样品色谱峰丰富,峰型较好,能更全面地反映药材信息;考察了不同样品提取方式(超声、回流)和提取时间(30、45、60及90 min),结果表明超声与回流各成分含有量无明显差异,提取45 min后样品有效成分含量较为接近,出于操作简便及提取效率的考虑,最终选定超声提取45 min;考察多种流动相体系梯度洗脱结果(乙腈-0.1%冰醋酸溶液、乙腈-0.1%磷酸溶液、乙腈-水、甲醇-0.1%冰醋酸溶液、甲醇-0.1%磷酸溶液及甲醇-水),结果表明乙腈-0.1%磷酸溶液分离度较好、出峰快、色谱峰较多;考察了不同柱温(25、30、35及40 ℃),30 ℃时出峰时间合适,峰型良好;采用DAD检测器收集200~400 nm全波长扫描下的紫外吸收图谱,观察3D图谱发现254 nm下各测定成分色谱峰的响应均较高,分离效果良好,基线稳定。因此,最终实验条件定为采用70%甲醇为提取溶剂,超声提取45 min,采用乙腈-0.1%磷酸溶液为流动相、检测柱温为30 ℃,检测波长为254 nm。
中药材化学成分复杂,本实验前期采用超高效液相色谱—离子阱飞行时间高分辨质谱联用仪,得到赤胫散药材中化合物分子量信息,将碎片离子信息与现有文献比对[27-29],经初步鉴定后与对照品紫外吸收光谱比对,进行化学成分的确定。本实验采用HPLC法建立了赤胫散的指纹图谱,标定了19个共有峰,指认出3种有效成分并进行含量测定。17批赤胫散样品与对照图谱相似度为0.768~0.997,各化学成分相对保留时间较为均一,但相对峰面积RSD值较大,不同批次间的同一成分含量也有所不同,说明样品间化学成分相似,但含量差异较大。通过聚类分析、主成分分析及正交偏最小二乘判别分析等化学计量学方法[23-26]对17批赤胫散指纹图谱进行综合分析,建立样品分类模型,发现了明显聚类趋势,将17批样品聚为2类,如鞣花酸含量为1.180 4~15.212 8mg/g,Ⅰ类样品鞣花酸含量在5.655 5mg/g以上且均高于Ⅱ类;以VIP值>1作为标准,筛选出10种引起赤胫散质量差异的主要标志成分,Ⅰ类样品该10种成分峰面积大多高于Ⅱ类;从药材粉末外观上看,Ⅰ类样品为棕黄色,Ⅱ类样品为棕红色,与聚类分析结果一致。由于其均为野生,药材产地、生长环境、海拔高度、储藏条件及时间等多种因素均可能导致药材化学成分降解或转化,为保证赤胫散质量,用药过程中应规范药材来源、采收时间、储藏条件并关注该10种成分的质量变化,可将其作为药材质量评价指标。
综上所述,本研究建立了赤胫散HPLC指纹图谱并同时测定3种有效成分含量,方法快速、操作简便、分离度好、化学成分信息丰富,指纹图谱定性结合有效成分定量,可弥补现有标准与含量测定方法的不足,对赤胫散药材进行整体性评价并反映其内在质量,为进一步质量控制及药材开发提供参考。
