Identification of four novel prognosis biomarkers and potential therapeutic drugs for human colorectal cancer by bioinformatics analysis
2021-03-13ZhenSunChenLiuStevenCheng
Zhen Sun, Chen Liu, Steven Y. Cheng,3,✉
1Department of Medical Genetics, 2Department of Pathology and Pathophysiology, 3Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Cancer Personalized Medicine, Nanjing Medical University, Nanjing, Jiangsu 211166, China.
Abstract Colorectal cancer (CRC) is one of the most deadly cancers in the world with few reliable biomarkers that have been selected into clinical guidelines for prognosis of CRC patients. In this study, mRNA microarray datasets GSE113513, GSE21510, GSE44076, and GSE32323 were obtained from the Gene Expression Omnibus (GEO)and analyzed with bioinformatics to identify hub genes in CRC development. Differentially expressed genes(DEGs) were analyzed using the GEO2R tool. Gene ontology (GO) and KEGG analyses were performed through the DAVID database. STRING database and Cytoscape software were used to construct a protein-protein interaction (PPI) network and identify key modules and hub genes. Survival analyses of the DEGs were performed on GEPIA database. The Connectivity Map database was used to screen potential drugs. A total of 865 DEGs were identified, including 374 upregulated and 491 downregulated genes. These DEGs were mainly associated with metabolic pathways, pathways in cancer, cell cycle and so on. The PPI network was identified with 863 nodes and 5817 edges. Survival analysis revealed that HMMR, PAICS, ETFDH, and SCG2 were significantly associated with overall survival of CRC patients. And blebbistatin and sulconazole were identified as candidate drugs. In conclusion, our study found four hub genes involved in CRC, which may provide novel potential biomarkers for CRC prognosis, and two potential candidate drugs for CRC.
Keywords: colorectal cancer, Gene Expression Omnibus, biomarkers, bioinformatics analysis
Introduction
Nowadays, colorectal cancer (CRC) is one of the most deadly cancers and almost 900 000 CRC-related deaths were reported each year in the world[1]. With the understanding of pathophysiology of the disease,different treatment options to improve the survival rates of CRC patients have been developed in the world. The 5-year survival rate of CRC patients was>90% when the patients were diagnosed at early stages[2]. However, due to lacking early detection methods, many CRC patients were diagnosed at an advanced stage or in the metastasis status. And the 5-year survival rate for those diagnosed with metastasis was at approximately 12%[3]. Recently, a new kind of analysis method has been used to identify the differential expression genes between CRC and normal tissues based on the high-throughput sequencing platforms, such as microarrays. This is a promising tool with extensive clinical applications,including molecular diagnosis, prognosis prediction,new drug targets discovery, etc[4-6]. Furthermore,microarray assay combining bioinformatics analysis made it possible to analyze the gene expression on mRNA level in CRC progression. For example,several studies have used this method to identify key genes in CRC development through comparing with normal samples, and showed that the key genes were involved in different signal pathways, biological processes, and molecular functions[7-12].
However, with a relatively limited degree of overlap, we still can not find reliable biomarkers or drug targets. Therefore, the discovery of novel biomarkers for early detection and prognosis prediction of CRC is urgently required.
In the present study, we targeted to find key genes to develop novel biomarkers or drug targets for CRC.Therefore, we chose four Gene Expression Omnibus(GEO) datasets, GSE113513, GSE21510, GSE44076,and GSE32323, and used bioinformatics methods to screen the significant differentially expressed genes (DEGs) between CRC tissues and normal tissues. Gene ontology (GO) and KEGG Pathway analyses were used to find the biological roles of these DEGs through DAVID database. Furthermore, the PPI network of DEGs was constructed and key modules or hub genes were selected with Molecular Complex Detection (MCODE) plugin of Cytoscape software.And the clinical significance was validated by GEPIA database. Finally, small active candidate molecules were identified to develop new drugs through Connectivity Map (CMap) database. In brief, we found four hub genes involved in CRC, which may provide novel potential biomarkers for CRC prognosis, and two potential candidate drugs for CRC.
Materials and methods
Data resources
To explore the differential gene expression profiles between CRC and normal tissues, we searched the NCBI-GEO database to collect enough and adequate tissues. A total of 4 GEO datasets were selected,including GSE113513, GSE21510, GSE44076, and GSE32323. These mRNA profiles were based on platform GPL15207 (GSE113513), GPL570(GSE21510 and GSE32323), and GPL13667(GSE44076). A total of 253 CRC samples and 203 normal samples were chosen for this study, including 14 pairs of cancer and normal samples in GSE113513,124 CRC samples and 24 normal samples in GSE21510, 98 pairs of cancer and normal samples plus 50 healthy donor tissues in GSE44076, and 17 pairs of cancer and normal samples in GSE32323.
Identification of DEGs and data preprocessing
To identify the DEGs, we used the NCBI-GEO2R online tool to analyze these datasets. Subsequently,adjustedP-value <0.05 and |log2(fold change)| >1 were set as the cutoff criteria to screen the significant DEGs of each dataset. Finally, Venn diagrams were performed to get the overlap significant DEGs of the 4 datasets.
GO and KEGG pathway analyses of DEGs
To find the biological functional roles of DEGs, GO and KEGG pathway analyses were performed through DAVID database. Significant results of molecular function (MF), biological process (BP), cellular component (CC), and biological pathways were selected withP-value <0.05.
PPI network construction and module analysis
The DEGs profiles were submitted to STRING database for exploring their potential interactions.The interactions with a combined score >0.4 were considered significant. Subsequently, the interaction files were downloaded and imported into Cytoscape software to construct the PPI network. The MCODE plugin was used to find key modules of the whole PPI network with a degree cutoff=2, node score cutoff=0.2, K-core=2, and max depth=100. The hub genes were then selected with connectivity degree >10. Furthermore, KEGG pathway analyses of the significant modules were performed withP-value <0.05.
Analysis and validation of hub genes
To verify the hub genes we found, we used GEPIA database to analyze their expression and clinical prognostic information in 270 CRC patients. And the survival curve, stage analysis and box plot were performed to show the clinical implications of hub genes.
Identification of small molecules
To find potential small active molecules to develop new drugs for treating CRC, we uploaded DEGs probe profiles into the CMap database. This database can help to predict small molecules that induce or reverse gene expression signature with a score from-1 to 1. And the molecules which value from 0 closer to -1 were functioned as reversing the cancer cell status.
Results
Identification of DEGs in CRC
Analyzed with the GEO2R online tool, a total of 1763, 4411, 2428, and 2276 DEGs were extracted from GSE113513, GSE21510, GSE44076, and GSE32323, respectively, using adjustedP-value <0.05 and |log2(fold change)| >1 as cutoff criteria. The volcano plots of DEGs in each dataset were shown inFig. 1A. And the Venn diagrams showed that 865 overlap DEGs were identified from these four datasets, including 374 significantly upregulated genes and 491 downregulated genes (Fig. 1BandSupplementary Table 1, available online).
Enrichment analysis of DEGs
To explore the biological functional roles of the overlap DEGs, GO and KEGG analyses were performed on DAVID database. And the top 20 terms were listed in the charts (Fig. 2A-DandSupplementary Table 2, available online). The GO analysis results consist of three functional categories,including BP, CC, and MF. In the BP group, DEGs were mainly enriched in cell proliferation (Fig. 2A).In the CC group, DEGs were enriched in cytoplasm(Fig. 2B). And in the MF group, DEGs were enriched in protein binding (Fig. 2C). KEGG pathway analysis showed that DEGs were enriched in metabolic pathways, pathways in cancer and cell cycle (Fig. 2D).The details of the top 20 terms were listed inSupplementary Table 2.
PPI network construction and module analysis
Using the STRING online database and Cytoscape software, a total of 865 DEGs were filtered into the PPI network complex, containing 863 nodes and 5817 edges (Fig. 3). Based on degree scores using the MCODE plugin, two key modules were detected from the whole PPI network complex. Module 1 contained 61 nodes and 1648 edges, and DEGs were enriched in cell cycle, oocyte meiosis, progesterone-mediated oocyte maturation, DNA replication and p53 signaling pathway (Fig. 4AandB). Module 2 had 55 nodes and 625 edges, and these DEGs were enriched in chemokine signaling pathway, ribosome biogenesis in eukaryotes, cytokine-cytokine receptor interaction,pathways in cancer, purine metabolism, RNA polymerase, retrograde endocannabinoid signaling,TNF signaling pathway, legionellosis, regulation of lipolysis in adipocytes, NOD-like receptor signaling pathway, cytosolic DNA-sensing pathway and gastric acid secretion (Fig. 4CandD). Additionally, the top 20 hub genes,CDK1,CCNB1,MYC,CCNA2,MAD2L1,AURKA,TOP2A,CDC6,UBE2C,CHEK1,RRM2,BUB1B,TTK,TRIP13,TPX2,BUB1,NCAPG,KIF2C,KIF23, andMCM4were identified with higher degrees of connectivity. These hub genes were enriched in cell cycle, progesterone-mediated oocyte maturation, oocyte meiosis, p53 signaling pathway,and HTLV-I infection (Fig. 4EandF).
Analysis and validation of hub genes
To validate the hub genes we got from this study,we uploaded the hub genes list into GEPIA database and explored the correlation between hub genes expression and the clinical characteristics of CRC. It was found thatHMMR,PAICS,ETFDH, andSCG2were significant DEGs in 270 CRC samples from GEPIA (Fig. 5A). And these four genes could represent the important prognostic biomarkers for predicting the survival of CRC patients (Fig. 5B).Meanwhile,PAICSandSCG2were related to the stages of CRC progression (Fig. 5C). The summaries of four hub genes were shown inTable 1.
Identification of related active small molecules
To search candidate small molecules for developing potential drugs to treat CRC, we uploaded DEGs probe profiles into the CMap database. And the predicted results were download and filtered with enrichment score <0 andP-value <0.05. The results were shown inTable 2. AndFig. 5Dlisted the top 20 small molecules with their enrichment scores andP-values. Therefore, these small molecules may be the targets to develop new drugs or therapies of CRC.Among these molecules, Blebbistatin and Sulconazole may be selected for new clinical trials.
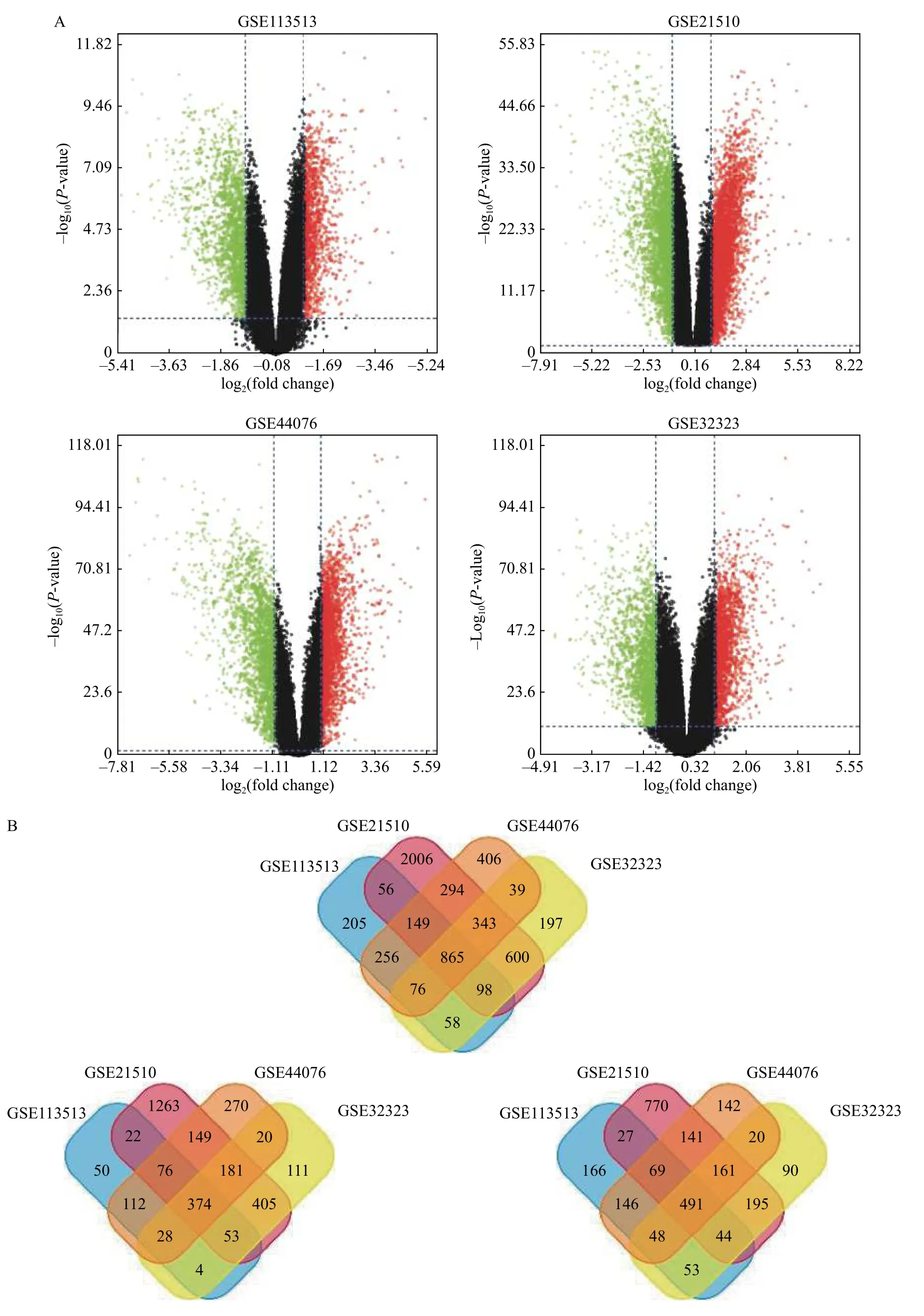
Fig. 1 Identification of the DEGs in CRC. A: Volcano plots of gene expression profiles between CRC and normal samples from GSE113513, GSE21510, GSE44076 and GSE32323. Red dots: significantly upregulated genes in CRC; Green dots: remarkably downregulated genes in CRC. Adjusted P value <0.05 and |log2(fold change)| >1 were considered as significant criteria. B: Venn diagrams show that 865 overlap DEGs were found through GEO2R in the four datasets, including 374 upregulated DEGs and 491 downregulated DEGs. DEGs: differentially expressed genes; CRC: colorectal cancer.
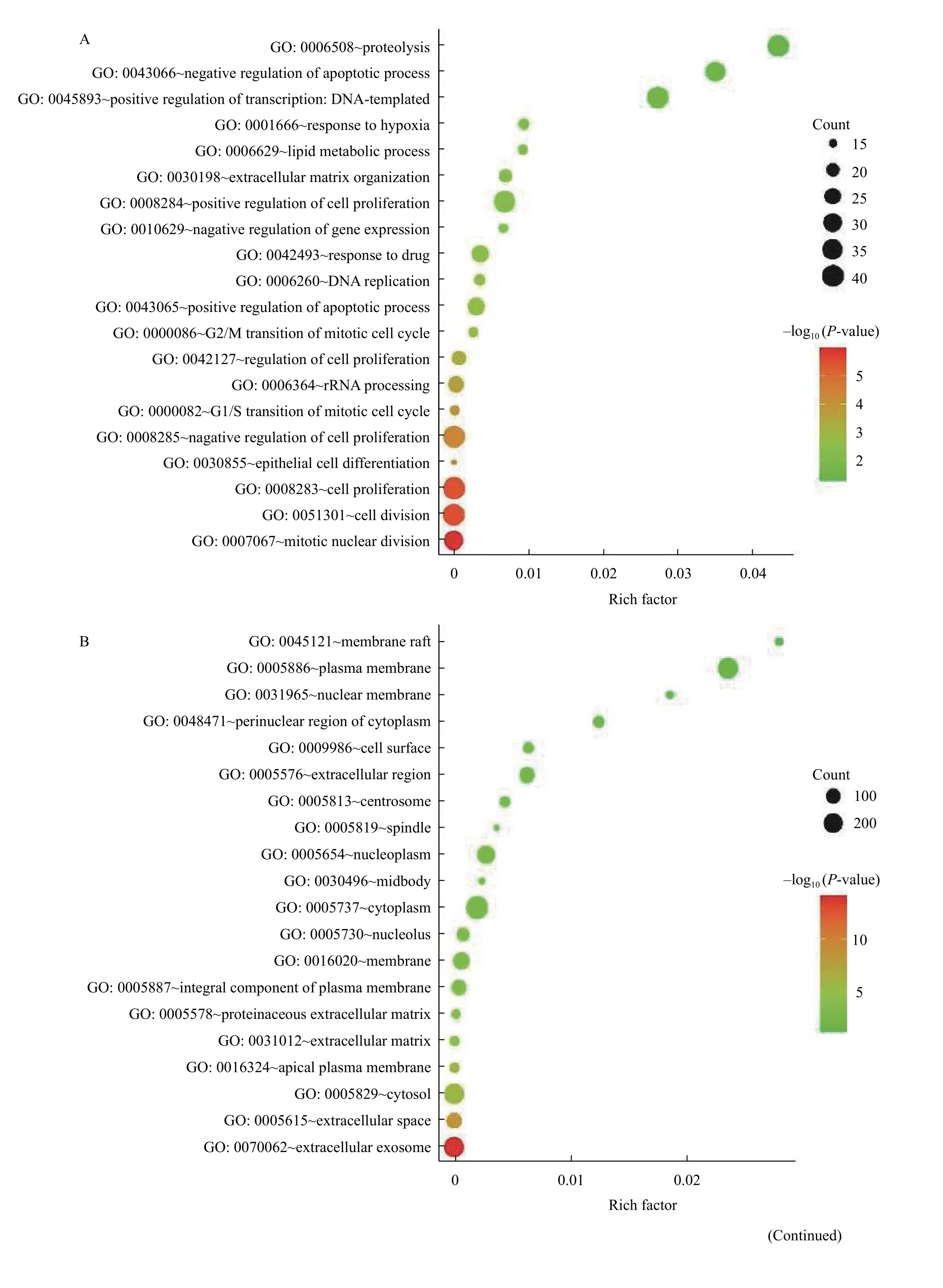
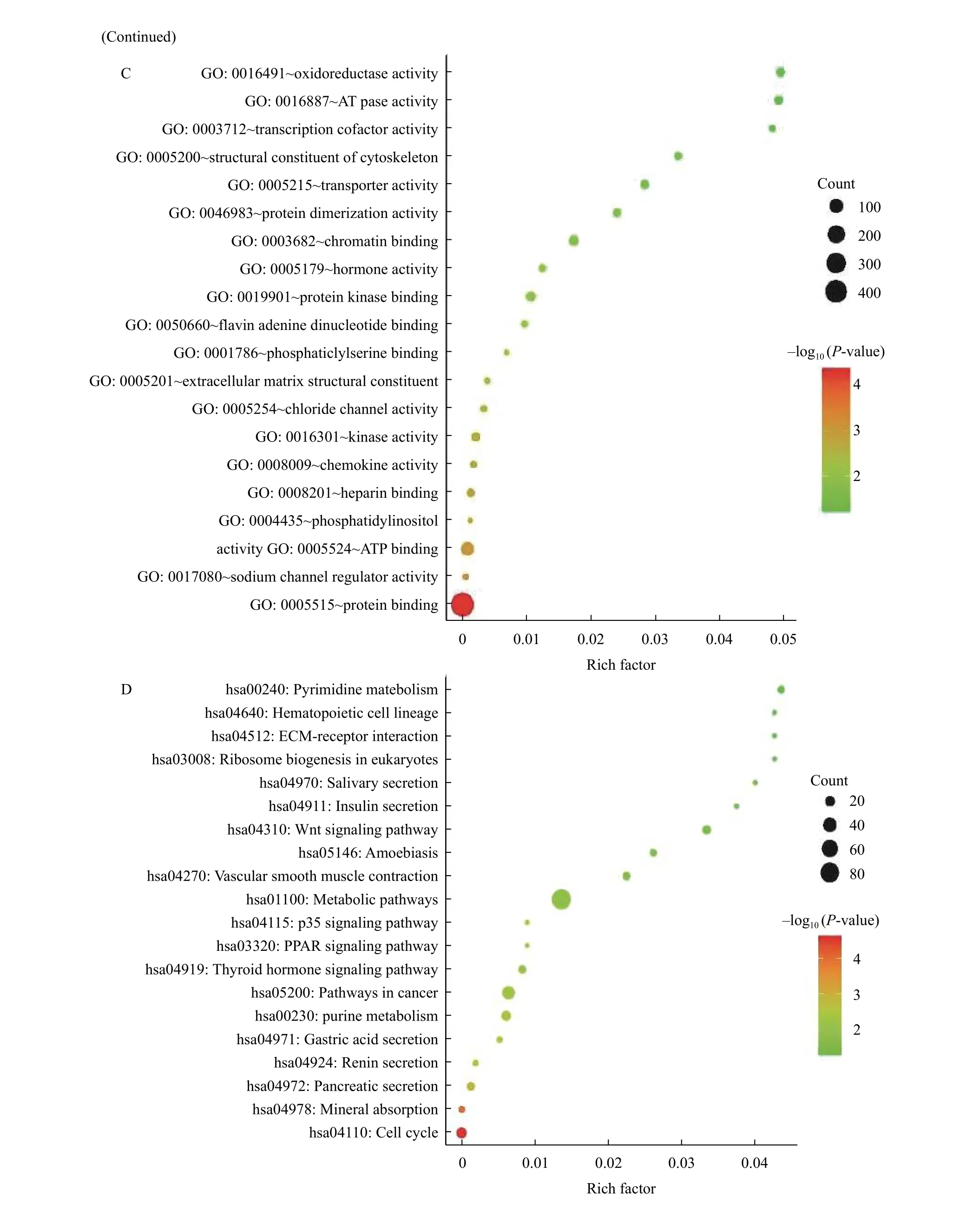
Fig. 2 GO and KEGG analysis of the overlap differentially expressed genes in colorectal cancer through DAVID online-tools.Top 20 terms of biological processes (A), cellular components (B), molecular functions (C), and KEGG signaling pathways (D) were shown in the charts, and P-value <0.05 was considered as selection criteria.

Fig. 3 PPI interaction network of the overlap differentially expressed genes by STRING database.
In summary, we chose GSE113513, GSE21510,GSE44076, and GSE32323 GEO datasets and found 865 significant DEGs between CRC tissues and normal tissues. Subsequently, the biological roles of these DEGs were confirmed with enrichment pathway analysis. Furthermore, the four hub genes,HMMR,PAICS,ETFDH, andSCG2were identified as important prognostic biomarkers for predicting the survival of CRC patients based on the GEPIA database. Finally, blebbistatin and sulconazole were picked out to develop new drugs through CMap database (Fig. 6).
Discussion
In our study, we chose four GEO datasets and used bioinformatics methods to get 865 DEGs (374 upregulated and 491 downregulated). KEGG pathway analysis showed that the key modules were mainly metabolic pathways, pathways in cancer, cell cycle,purine metabolism, pancreatic secretion, thyroid hormone signaling pathway and Wnt signaling pathway. The PPI network was constructed including 863 nodes and 5817 edges. The four hub genes,HMMR, PAICS, ETFDH, and SCG2 were remarkably related to the prognosis of patients. Furthermore, two small molecules, blebbistatin and sulconazole, also have been identified as potential candidates to develop new drugs.

Recently, findings about DEGs or molecular biomarkers of CRC have been increasingly reported.Based on integrated analysis of GSE32323,GSE74602, and GSE113513 datasets, and TCGA databases,CCL19,CXCL1,CXCL5,CXCL11,CXCL12,GNG4,INSL5,NMU,PYY, andSSTwere identified as hub genes. And 9 genes includingSLC4A4,NFE2L3,GLDN,PCOLCE2,TIMP1,CCL28,SCGB2A1,AXIN2, andMMP1was related to predicting overall survivals of CRC patients[8].Moreover,TOP2A,MAD2L1,CCNB1,CHEK1,CDC6, andUBE2Cwere indicated as hub genes, andTOP2A,MAD2L1,CDC6, andCHEK1may serve as prognostic biomarkers in CRC[10]. In addition,CEACAM7,SLC4A4,GCG, andCLCA1genes were associated with unfavorable prognosis in CRC[11].According to analysis of GEO datasets and survival analysis by GEPIA database,AURKA,CCNB1,CCNF, andEXO1were significantly associated with longer overall survival. Moreover, CMap predicted that DL-thiorphan, repaglinide, MS-275, and quinostatin have the potential to treat CRC[9]. In this study, we have identified four hub genes as new potential biomarkers to predict the prognosis of CRC patients and two new small molecules. Further studies are needed to develop new drugs to treat CRC.
Several studies have reported that these hub genes play important roles in cancer development. For instance,HMMRexpression level was remarkably correlated with the progression and prognosis of breast cancer[13], bladder cancer[14], prostate cancer[15-16], lung cancer[17-19], hepatocellular carcinoma(HCC)[20-22], and gastric cancer[23]. Furthermore,HMMRwas confirmed to maintain its oncogenic properties and resistance to chemotherapy through activating TGF-β/Smad-2 signaling pathway[24]. AndHMMRwas highly expressed in glioblastoma and related to support the self-renewal and tumorigenic potential of glioblastoma stem cells[25].PAICSwas also upregulated in several kinds of cancer tissues and it promotes cancer cells proliferation, migration, and invasion[26-31]. The expression level ofETFDHwas found significantly decreased in HCC tissues, and this low expression was related to poor overall survival in patients[32]. However, the role ofSCG2in cancer remains unclear.
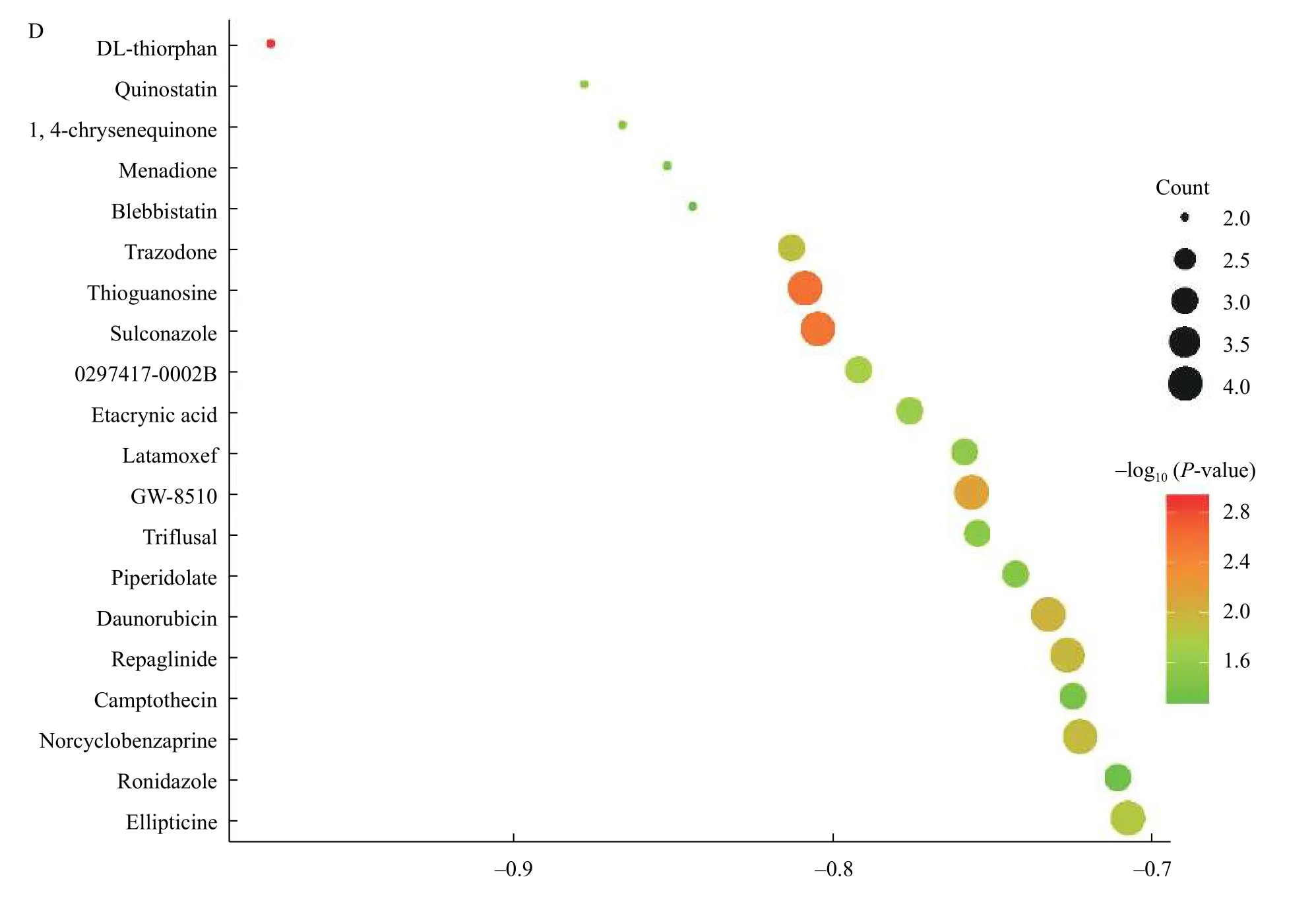
Fig. 5 Analysis and validation of hub genes and identification of related active small molecules. A and B: The expression level and prognostic value of four hub genes based on the GEPIA database. C: PAICS and SCG2 were related with the stages of CRC progression through GEPIA database. D: Top 20 potential small active molecules reverse DEG of CRC predicted by CMap database. TPM: transcripts per million.
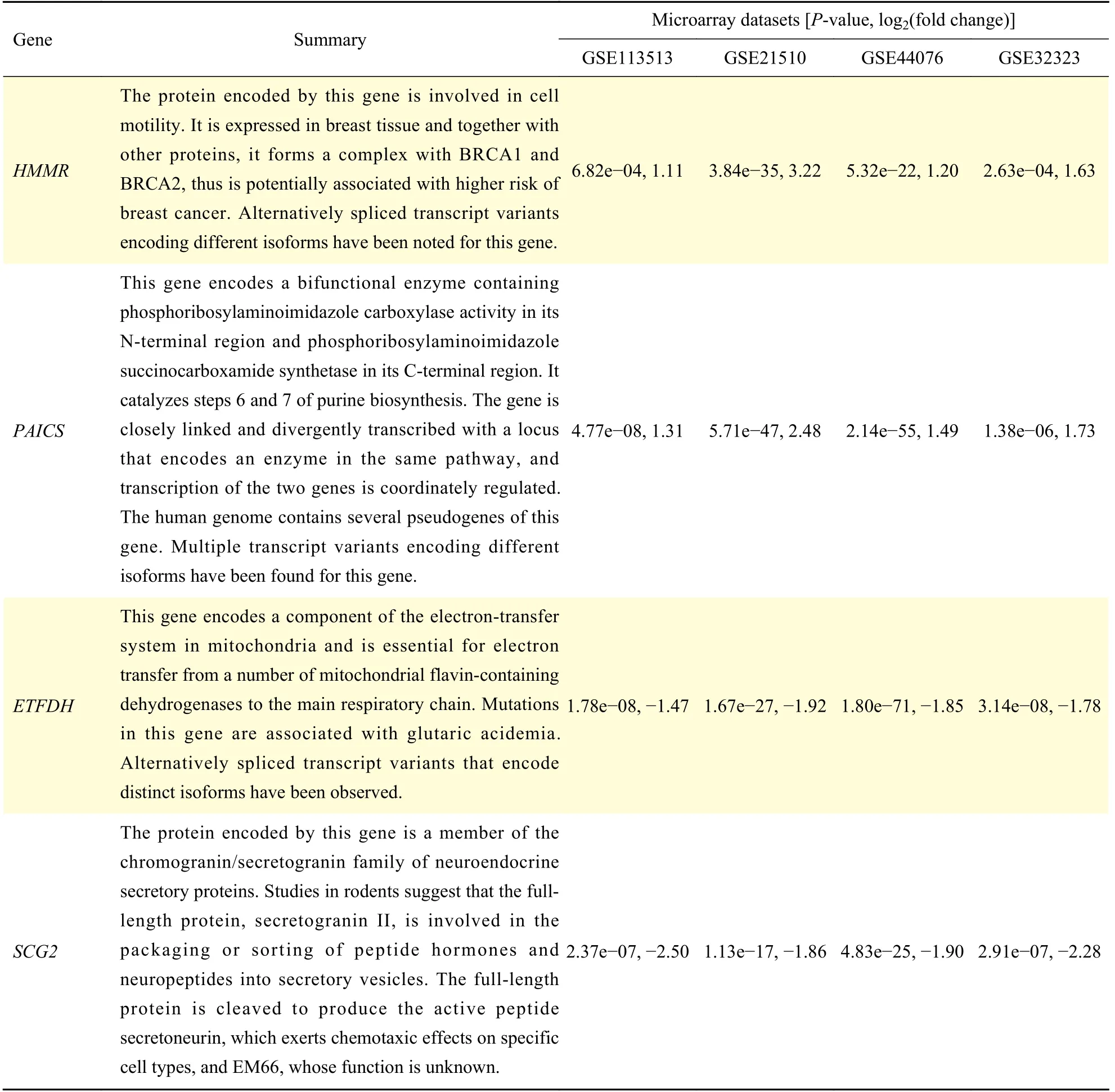
Table 1 Gene summaries of the four hub genes
In the present study, HMMR, PAICS, ETFDH, and SCG2 were significantly up or down regulated in CRC tissues compared with those in normal samples, and the survival rate of CRC patients was positively correlated with the expression of these genes. Besides,several small molecules with potential therapeutic efficacy were identified through bioinformatics analyses, including blebbistatin and sulconazole.Blebbistatin has been reported to inhibit cell migration and invasiveness of pancreatic adenocarcinoma[31], and decrease spreading and migration of breast cancer cells[33]. Moreover, blebbistatin has shown its antitumorigenic properties in HCC cells[34]. Another small molecule, sulconazole, also inhibited the proliferation and formation of breast cancer stem cells through blocking the NF-κB/IL-8 signaling pathway[35]. Although these two molecules have significant antitumor activity, their specific roles in CRC development need to be further clarified.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (No.81672748 and No. 81871936).
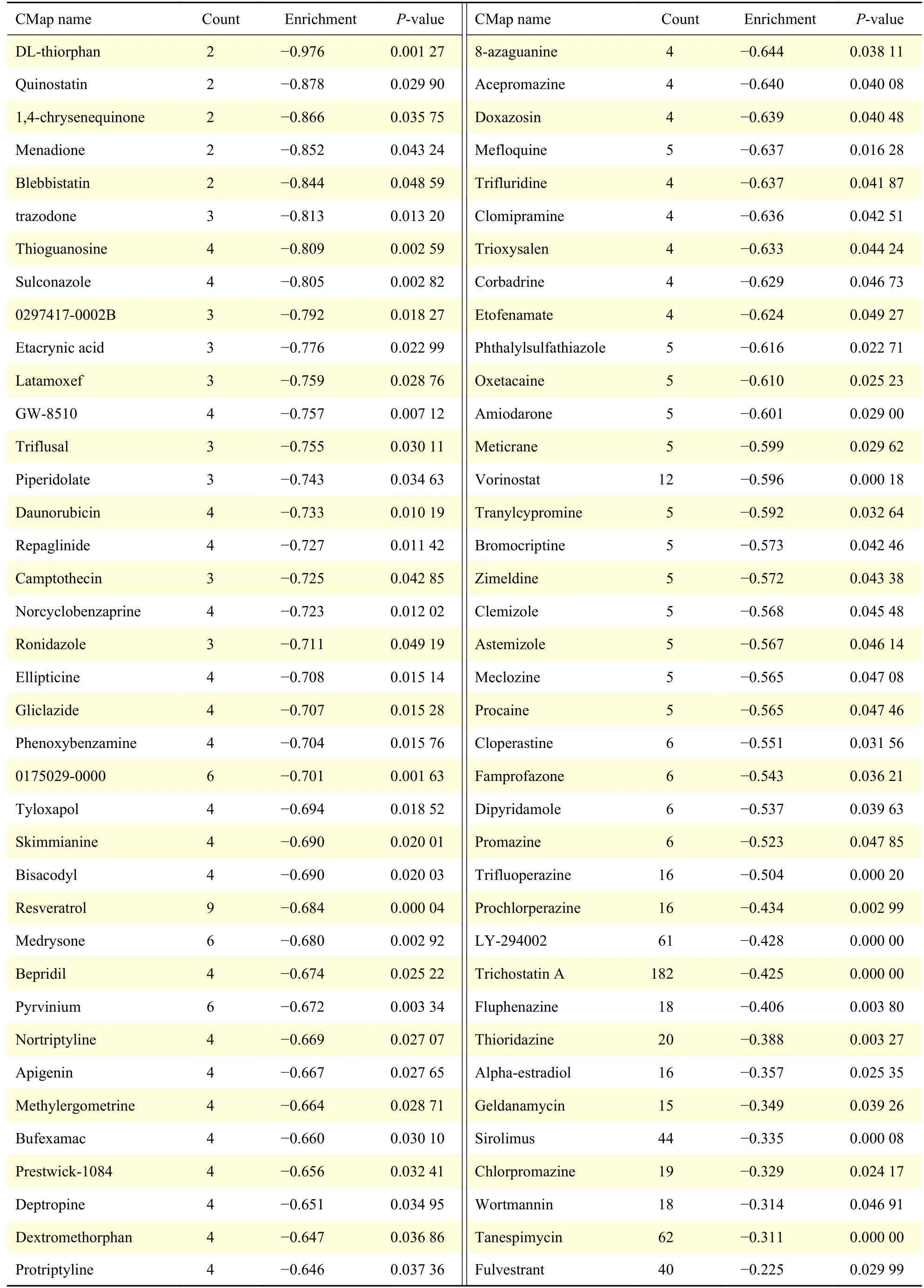
Table 2 The significant small active molecules that may reverse the DEGs of CRC predicted by CMap
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Contained local compression on peri-ascending aortic area for postoperative bleeding control: a case report
- Intrathecal hematoma and sacral radiculitis following repeat epidural blood patch
- Valproic acid therapy decreases serum 25-hydroxyvitamin D level in female infants and toddlers with epilepsy— a pilot longitudinal study
- The testis-specifically expressed gene Trim69 is not essential for fertility in mice
- α-ketoglutarate promotes the specialization of primordial germ cell-like cells through regulating epigenetic reprogramming
- A comparative genomics analysis of lung adenocarcinoma for Chinese population by using panel of recurrent mutations