A comparative genomics analysis of lung adenocarcinoma for Chinese population by using panel of recurrent mutations
2021-03-13WanlinLiMinWuQianqianWangKunXuFanLinQianghuWangRenhuaGuo
Wanlin Li, Min Wu, Qianqian Wang, Kun Xu, Fan Lin, Qianghu Wang, Renhua Guo,✉
1Department of Bioinformatics, Nanjing Medical University, Nanjing, Jiangsu 211166, China;
2Department of Oncology, the First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu 210029, China;3Department of Cell Biology, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu 211166,China;
4Jiangsu Key Lab of Cancer Biomarkers, Prevention and Treatment, Collaborative Innovation Center for Cancer Personalized Medicine, Nanjing, Jiangsu 211166, China.
Abstract
Keywords: lung adenocarcinoma, Chinese population, ethnic difference, genomic characteristics, targeted sequencing
Introduction
Lung cancer is the most prevalent cancer and the prime cause of cancer death both worldwide and in China[1-3], with a 5-year survival rate of lower than 20% according to the 2018 annual report[4], which suggests that lung cancer is still a huge threat to public health. Based on the classification standard of the World Health Organization, lung cancer can be divided into non-small cell lung cancer (NSCLC) and small cell lung cancer. Among the NSCLC, the lung adenocarcinoma (LUAD) accounts for approximately 63%[5], and is the most common subtype in nonsmokers, especially non-smoking Asian women[6-7].Despite substantial epidemiological statistics have shown that cigarette smoking and second-hand smoke exposure are the major risk factors of lung cancer[8-10],the specific pathogenesis and mechanism of lung cancer are still unknown.
Extensive genomic studies have been conducted to identify genetic variants and recurrent somatic mutations involved in the development of lung cancer,although most patients were recruited from western countries. Therefore, the genomic characteristics of the Chinese population need to be elucidated. Previous studies on Chinese patients have discovered 14 susceptibility loci that are specific to the Chinese population, of which 5 loci (rs4809957, rs2895680,rs247008, rs2736100, and rs9439519) are associated with smoking dose[11]. In addition, gene mutation rates varied between diverse ethnic populations[12]. For instance, epithelial growth factor receptor (EGFR) is identified as a driver gene of LUAD, and alters in 50%-60% of Asians and 15%-20% of Caucasians[13].The most common L858R mutation located in the kinase domain of EGFR, which is sensitive to EGFR tyrosine kinase inhibitors (EGFR-TKI), is observed to be more frequent in Asian LUAD than in Caucasian groups, which indicates the Chinese will benefit more from EGFR-TKI treatment[14]. Other genes likeKRAS,TP53, NF1,andKEAP1also present differential mutation rates in Chinese and Caucasian samples[12].Taken together, these results demonstrated that ethnicity plays a pivotal role in the detected frequency of genetic markers, and the genomic features of Chinese LUAD need to be further understood.
Targeted sequencing is a powerful technology to detect mutations occurring in interested genes owing to its higher coverage in genomic loci. Moreover,targeted sequencing enables the estimation of panelbased tumor mutational burden (pTMB).
In this study, we implemented targeted sequencing on 66 Chinese LUAD patients and compared their samples with 162 Caucasian LUAD samples acquired from The Cancer Genome Atlas (TCGA). We revealed that different genomic alteration profiles and mutation patterns exist in Chinese LUAD and Caucasian LUAD. Moreover, we identified novel driver genesGNASandJAK1that are specific to Chinese LUAD, which may contribute to the diagnosis and treatment of LUAD.
Materials and methods
Sample collection
We collected a total of 66 formalin-fixed paraffin embedded (FFPE) LUAD specimens from the First Affiliated Hospital of Nanjing Medical University during March 2015 and May 2018. Afterward, tumor tissues and matched peripheral blood of patients were sent to perform targeted DNA sequencing.
This study was approved by the Ethics Committee of Nanjing Medical University, and all patients signed informed consent for the research. Besides, all clinical data and samples were received anonymously.
Acquisition of public data
For comparative analysis, clinical information and mutational data of 173 Caucasian LUAD samples were downloaded from the Broad Firehose Infrastructure(http://www.broadinstitute.org/cancer/cga/Firehose),and of them, 11 samples which harbored only silent mutations were excluded for further analysis. Detailed information of 162 samples are shown inTable 1.
DNA extraction
DNA was extracted from FFPE samples using QIAamp DNA FFPE Tissue Kit (Qiagen, Germany,Cat. #: 56404), and from peripheral blood samples using QIAamp DNA Blood Mini Kit (Qiagen,Germany, Cat. #: 51104). Afterward, DNA was quantified by dsDNA HS Assay Kit and Qubit 3.0(Thermo Fisher, USA, Cat. #: Q32851), and was broken into fragments of 350 bp by Covaris M220 ultrasound system, followed by purification using Agencourt AMPure XP beads (Beckman Coulter,Canada, Cat. #: A63881).
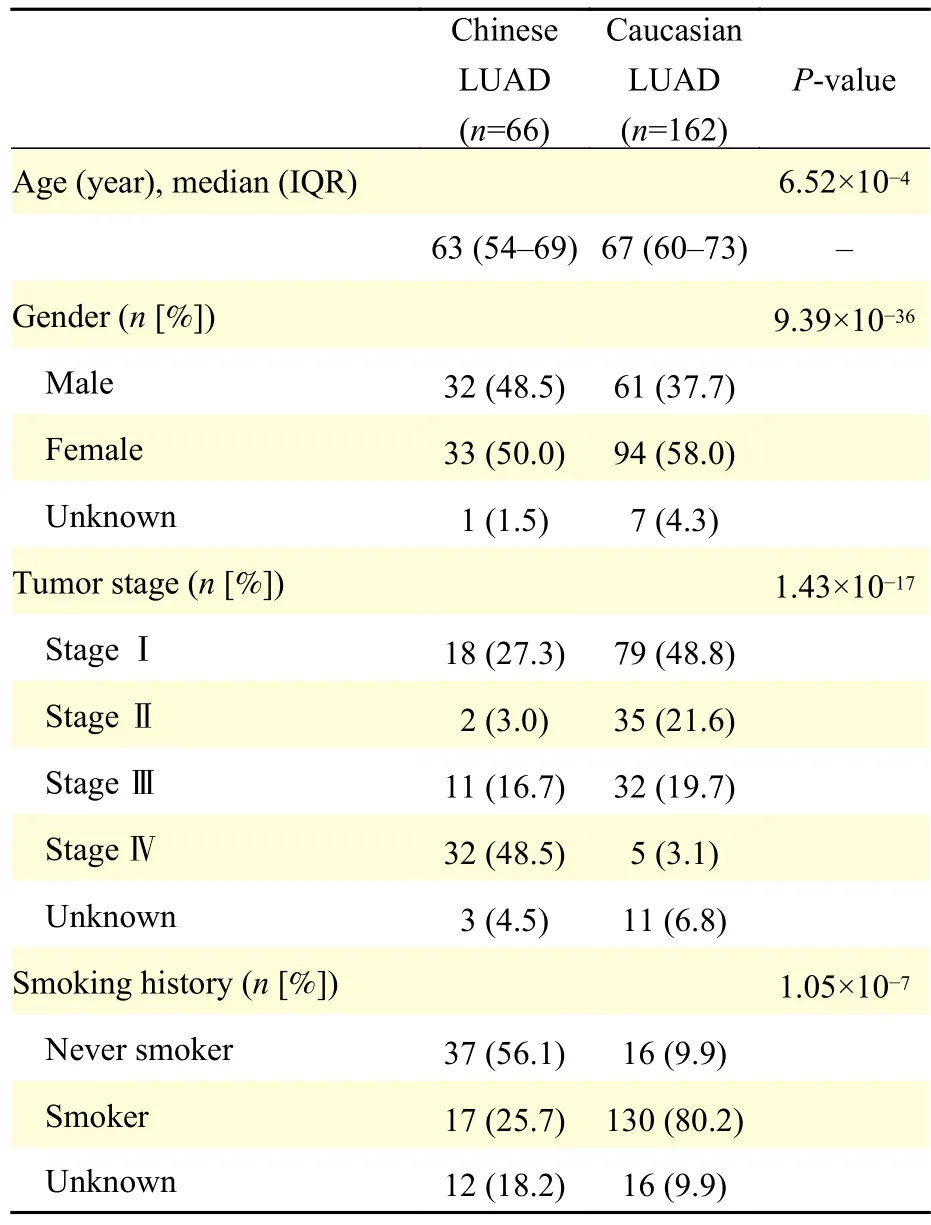
Table 1 Clinical information of LUAD patients
Library construction and targeted sequencing
DNA library was prepared using the KAPA Hyper Library Preparation kit (KAPA Biosystems, USA,Cat. #: KK8500), and targeted capture was performed by xGen Lockdown Reagents and customized gene probe (Integrated DNA Technologies, USA) and amplifiedviaKAPA HiFi HotStart ReadyMix (KAPA Biosystems, Cat. #: KK2602). The final libraries were quantitated using KAPA Library Quantification kit(KAPA Biosystems, Cat. #: KK4824) by qPCR, and the distribution of fragments was determined by Bioanalyzer 2100 (Agilent Technologies, USA).Finally, the 150 bp paired-end sequencing reads produced by HiSeq4000 (Illumina, USA) genome sequencer were obtained.
Processing of sequencing data
To achieve a higher coverage depth of interested genes, we performed targeted sequencing on 66 Chinese LUAD patients, of which 21 samples were sequenced by Geneseeq Prime panel (425 cancerrelated genes) and 45 samples sequenced by Gene+OncoD panel (1021 tumor-associated genes). The 425-gene panel detected 124 mutant genes, the 1021-gene panel detected 316 mutant genes, and TCGA whole-exome sequencing (WES) detected 12 290 mutant genes. We retained genes that were identified by all three datasets and obtained 68 genes. Finally,these 68 mutant genes were applied to subsequent analysis. The 68 gene symbols were listed inSupplementary Table 1(available online).
The quality control of raw sequenced reads was performed by FastQC (version 0.11.8), and most reads were found with a Phred score of more than 30. Then clean reads were mapped to human reference genome hg19 by Burrows-Wheller Aligner (BWA-MEM)(version 0.7.17)[15]. Duplicated reads were marked out and base quality scores were recalibrated by MarkDuplicates and BaseRecalibrator tool in the Genome Analysis Toolkit (GATK) (version 4.0.8.1)[16],and somatic variations (somatic single-nucleotide variations and insertion/deletion) were detected by Mutect2[17]. The obtained Variant Call Format (VCF)results were filtered by FilterMutectCalls and annotated by ANNOVAR (version 2018Apr16). All the figures were completed by R packages ggplot2(version 3.2.1)[18], G3viz (version 1.1.2)[19]and maftools (version 2.2.10)[3,20].
Calculation of convergent distribution index
We defined convergent distribution index (CDI) to measure the convergent level of mutation distribution[21]. The CDI was calculated as below:
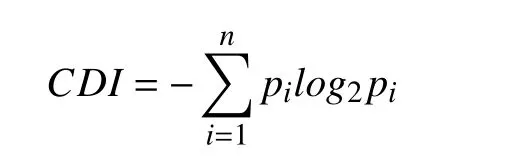
nrepresented the number of mutation loci of a specific gene, andpidenoted the occurrence probability of mutation at sitei, namely the ratio of mutations at siteito the total mutations on the gene. A lower CDI value indicated a more convergent mutation distribution in this study.
Statistical analysis
Wilcoxon rank-sum test was applied to continuous data when comparing the statistical differences between groups. Fisher's exact test was used to access the mutation distribution of Chinese LUAD and Caucasian LUAD. Shannon entropy was used to measure the convergent level of mutation distribution.Pearson correlation coefficient was calculated to measure the correlation between the two groups.
Results
The genomic variation landscape in Chinese and Caucasian LUAD
To comprehensively present the genomic alteration profile of LUAD patients from China and TCGA, we included 66 Chinese samples (32 males and 33 females, aged from 34 to 87 years old) and 162 Caucasian patients (61 males and 94 females, aged from 42 to 85 years old). As shown inTable 1, 25.7%(17/66) and 80.2% (130/162) of smokers were contained in the Chinese and Caucasian cohort respectively. Overall, Chinese LUAD (5 somatic mutations per sample) harbors more mutations than Caucasian cases (3 somatic mutations per sample)(Wilcoxon rank-sum test,P=0.0017) (Fig. 1A). Of these genomic alterations, missense mutations were the most common type in both cohorts (Chinese:68.3% [270/395]vs. Caucasian: 72.6% [461/635],P=0.16), which was consistent with previous studies.In addition, more in-frame indels are observed in Chinese cohort (2.0% [8/395]vs. 0.2% [1/635],P=2.67×10-3), while frame-shift insertions were more frequent in Caucasian cohort (1.3% [5/395]vs. 3.0%[19/635],P=0.089) (Fig. 1B). Recent studies revealed that in-frame indels more frequently occur in oncogenes to cause gain-of-function[22], while the high load of frame-shift indels was related to a better survival[23], which following the fact that Chinese LUAD were mainly composed of advanced-stage patients, and Caucasian LUAD mainly early-stage patients (Table 1).
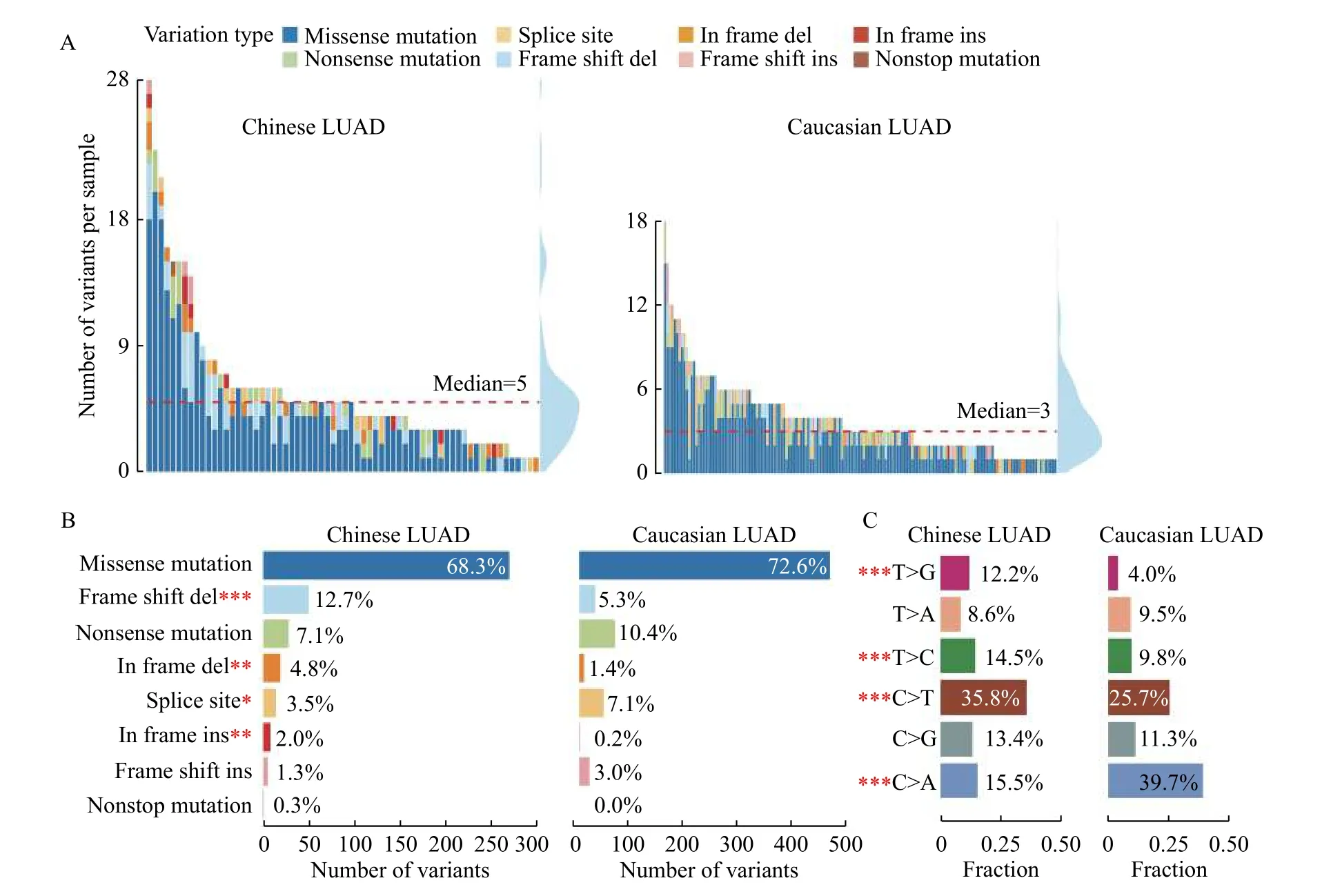
Fig. 1 Genomic variation landscape of Chinese and Caucasian LUAD patients. A: Summary of different types of variations in Chinese and Caucasian samples. Each bar represents a sample, and the colors represent variation types as described in the legend. The density plot in the right shows the respective distribution of mutation counts in each population. B: Statistics of variant types among Chinese and Caucasian patients. C: Composition of point mutations in Chinese and Caucasian patients. LUAD: lung adenocarcinoma. ***P<0.001, **P<0.01, *P<0.05.
We further compared the distribution of single nucleotide variants (SNVs) between Chinese and Caucasian. SNVs including 6 different mutation forms can be classified into transition (Ti) and transversion(Tv). Our results revealed that transition events from C to T are prevalent (35.8%, 330/922) in Chinese patients, which were induced by ultraviolet light[24].Studies revealed that long fixation time of tissues can trigger deamination and thus increase C/G>T/A mutations[25], so whether the more frequent C>T mutations in Chinese LUAD were caused by differences in populations or FFPE tissues needed more samples to verify. While transversion events from C to A were frequently observed (39.7%,256/645) in Caucasian patients (Fig. 1C). Chinese cohort have a higher Ti/Tv ratio than its counterpart(0.97 [454/468]vs. 0.59 [239/406],P=1.94×10-6). It was noteworthy that the cytosine-adenine (C>A)transversion in Caucasian is more frequently detected than in Chinese (Fig. 1C), which can be explained by a higher proportion of patients with smoking history in Caucasian cohort ( 80.2%vs. 25.7%,P=1.05×10-7)according to the clinical statistics (Table 1), because cytosine to adenine nucleotide transversions had been reported as a smoking-associated signature in many studies[26-27].
Overall, our mutational analysis demonstrated that missense mutations were ubiquitous in both cohorts,and C>A transversions were more frequently detected in Caucasian samples owing to the smoking behavior,while in-frame indels were more frequent in Chinese LUAD patients.
Comparison of mutation rate between Chinese and Caucasian LUAD
To further explore the somatic mutational characteristics of Chinese and Caucasian LUAD patients, we compared the mutation rates of 68 genes(Supplementary Table 1) in corresponding populations.Our results displayed that the two cohorts have different mutation profiles. The most common mutations in Chinese patients wereEGFR(66.7%,44/66) andTP53(54.5%, 36/66), and in Caucasian patients wereTP53(48.1%, 78/162) andKRAS(34.6%, 56/162) (Supplementary Fig. 1, available online).
EGFRhas been one of the most common mutations in LUAD patients, and accumulating evidence revealed that the incidence of EGFR mutations was higher in Asians than in Caucasians. As shown inSupplementary Fig. 1, the frequency ofEGFRmutations in Chinese patients is significantly higher than in Caucasian patients (66.7%vs. 15.4%,P=1.08×10-13). As a result, the Chinese can benefit more from EGFR-TKI treatments, which can provide effective control of tumor progression and prolong the overall survival ofEGFRmutant LUAD patients. This data sufficiently demonstrated the importance of preciseEGFRmutation detection to the treatments of Chinese LUAD patients.
On the other hand, the frequency ofKRASmutations in Caucasian samples was relatively higher than in Chinese samples (34.6%vs. 12.1%,P=5.6×10-4), which was consistent with previous results that theKRASmutation rate in European and American LUAD patients was about 15% to 30% and 10% to 15% in East Asian LUAD populations[6].Moreover, the fact thatKRASmutations were associated with tobacco consumption also leads to the increase ofKRASmutation in Caucasians[28].
Other genes likeBRD4(10.6%vs. 1.2%,P=2.8×10-3),CREBBP(15.2%vs. 4.3%,P=9.70×10-3),PALB2(10.6%vs. 1.2%,P=2.84×10-3),NSD1(10.6%vs. 1.2%,P=2.84×10-3), andEP300(10.6%vs. 1.2%,P=2.84×10-3) tended to mutate in Chinese population,while mutations located inKEAP1(6.1%vs. 18.5%,P=2.26×10-2) tended to occur in Caucasian samples(Supplementary Fig. 1andSupplementary Table 1).
Taken together, our results suggested that the tumor suppressor geneTP53universally mutates in LUAD patients, while the mutation rates ofEGFRas well as other 6 genes were ethnic dependent, andKRASis cigarette associated.
Identification of candidate driver mutations in Chinese LUAD using ultra-deep targeted sequencing
Driver mutations are defined as somatic alterations that could trigger tumorigenesis and generally undergo positive selection during the progression of cancer,thus displaying higher mutation rates than background mutations[29]. Given the considerable difference of genomic features induced by race, we identified potential driver mutations in Chinese and Caucasian LUAD. Alterations that occurred inKRASandEGFRwere the common driver mutations in both cohorts(Fig. 2AandB), which was consistent with previous reports that somatic mutations inKRASandEGFRcould initiate tumor[6]. Functional mutations inKRASandEGFRwere generally mutually exclusive (Fig. 2CandD), and co-existence of them was responsible for the resistance toEGFRinhibitors[2].
In addition,GNASandJAK1are identified as potential driver genes of Chinese LUAD. Mutations inGNASare involved in gastrointestinal tumors and exist in 66% of intraductal papillary mucinous neoplasm. While the mutation rate ofGNASin lung adenocarcinomas was much lower, 7.6% of Chinese patients harborGNASmutations in our cohort(Supplementary Fig. 1). Studies had shown thatGNASalterations are concurrent with the Raf/Ras pathway mutation[30].GNASmutations usually cooccur withSTAG2andCREBBPin Caucasian cohort(Fig. 2D).JAK1is a tyrosine kinase protein belonging to the Janus (JAK) family, which plays a crucial role in tumor-promoting inflammation[31-32], and alters in 10.6% of Chinese LUAD (Caucasian LUAD: 3.7%)along withNSD1mutation (Fig. 2C).
Collectively, apart from broadly discussed driver mutationsEGFRandKRAS, we additionally identifiedGNASandJAK1as potential driver mutations of Chinese LUAD.
Chinese LUADs present a convergent mutation distribution
To thoroughly elucidate the genomic difference, we compared the distribution of mutations located in identified driver genes. We defined a Shannon entropy-based indicator to measure the convergent level of mutations, termed as convergent distribution index (CDI). The value of CDI is negatively correlated with the concentrated distribution of mutations.
We selected outEGFR-mutant samples in Chinese(n=44) and Caucasian cohorts (n=25), and detected mutations located in primary domains of EGFR protein. As shown inFig. 3A, mutations ofEGFRmainly occur in the tyrosine kinase domain, and Chinese LUAD display concentrated distribution with a CDI value of 3.51 (Caucasian CDI: 3.88) (Fig. 3A).Likewise, we obtainedKRAS-mutant samples in Chinese (n=8) and Caucasian cohort (n=56), and predominant mutations occur in the Ras domain.KRASalterations in Caucasian LUAD tend to be more concentrated with a CDI value of 0.38 (Chinese CDI:1.75) (Fig. 3B). Moreover, other driver mutations located inGNAS(Chinese CDI: 1.25vs. Caucasian CDI: 2.73) (Fig. 3C) andJAK1(Chinese CDI: 1.66vs.Caucasian CDI: 2.81) also show a convergent trend in the Chinese cohort (Fig. 3Dand3E).
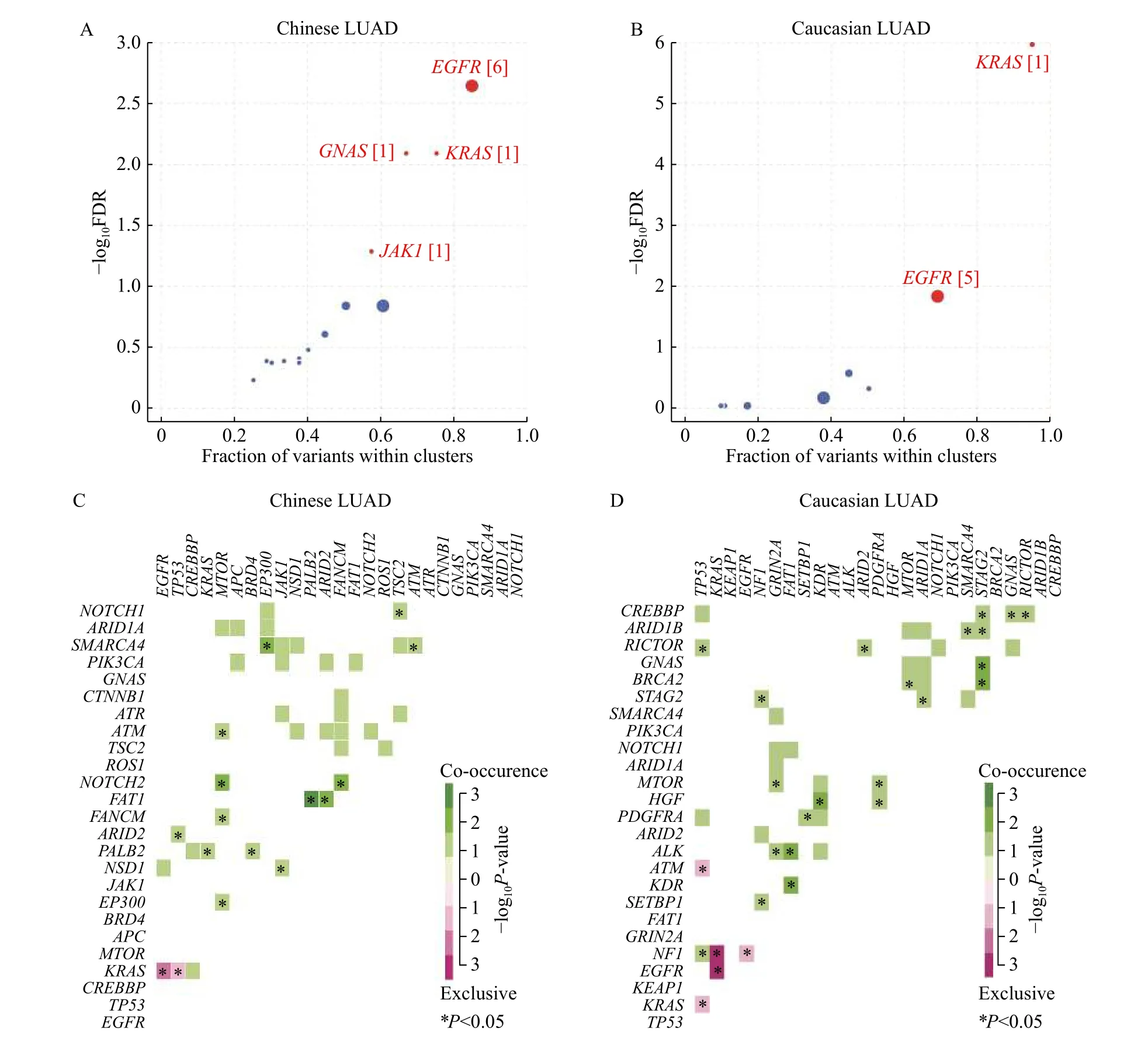
Fig. 2 Identified driver genes and co-occurrent or exclusive gene pairs. A and B: Scatter plots showing driver genes in Chinese (A) and Caucasian (B) cohorts (FDR<0.1). The size of the dot is positively associated with mutation clusters, and the number in bracket indicates the count of mutation clusters. C and D: Triangular matrix displaying the mutually exclusive and co-occurring gene pairs in Chinese (C) and Caucasian samples (D). Green indicates co-occurrent gene pairs, and red indicates exclusive gene pairs. LUAD: lung adenocarcinoma.
Further, we explored the CDI of other genes included in the targeted sequencing panel and found that CDI values of Chinese patients are significantly lower than those of Caucasian patients (Wilcoxon rank-sum test,P=0.012), which suggested a more clustered mutation distribution in Chinese LUAD patients (Fig. 3E). We checked the mutation distribution of 68 genes of the OncoSG dataset of 92 LUAD patients from Beijing[33]and found that the Beijing LUADs present a significantly convergent distribution than Caucasians (P=4.4×10-8), which is consistent with results from our dataset(Supplementary Fig. 2, available online).
In summary, our results revealed that mutations distribute more convergently in Chinese cohort than in its Caucasian counterpart.
TMB varies with the tumor stage of LUAD patients
Prior studies proved that NSCLC patients carrying higher tumor mutational burden (TMB) could benefit from the treatment of PD-1/PD-L1 inhibitors[34].Consequently, we explored the TMB among Chinese and Caucasian LUAD. We demonstrated that the panel-based TMB (pTMB) estimated by targeted sequencing is highly correlated with results by whole exome sequencing (R=0.82,P<0.001) (Supplementary Fig. 3B, available online). Therefore, it was reasonable to calculate TMBviatargeted sequencing[35].


Fig. 3 Convergent distribution of mutations in Chinese LUAD patients. A-D: Lollipop charts displaying the EGFR (A), KRAS (B),GNAS (C), and JAK1 (D), and mutations of Chinese and Caucasian patients. The x-axis shows the primary domains of EGFR protein, and the y-axis shows the number of mutations. The colors of circles represent mutation types, and the size of circles represents the count of mutations located in corresponding genomic loci. E: Shannon entropy of mutation distribution in 68 genes in Chinese and Caucasian samples. CDI:convergent distribution index; LUAD: lung adenocarcinoma. *P<0.05.
According to the clinical records, apart from 3 Chinese patients with the tumor stage information missing, Chinese cohort mainly consisted of stage Ⅳ(48.5%, 32/66) and stage I (27.3%, 18/66) patients,and patients of advanced stage (stage Ⅲ and Ⅳ)accounted for 65.2%. While the Caucasian cohort mainly consisted of stage I (48.8%, 79/162) and stageⅢ (19.7%, 32/162) patients, and patients of earlystage (stage Ⅰ and Ⅱ) accounted for 70.4% (Fig. 4A).On average, Chinese LUAD hold a higher pTMB(18.12 mutations/Mbvs. 12.48 mutations/Mb), which may be caused by the high proportion of advancedstage patients in the Chinese cohort (Fig. 4A). In addition, we noted that pTMB gradually increased with tumor progression in both populations, and higher pTMB is observed in Chinese patients with stage Ⅲ LUAD than in their Caucasian counterparts(Wilcoxon rank-sum test,P=0.01) (Fig. 4B), which suggests that advanced Chinese LUAD patients might have a better response to immunotherapy.
In addition, a previous study demonstrated that high TMB calculated by targeted sequencing was associated with improved clinical status in NSCLC patients, which indicated that pTMB could predict the response to immunotherapy[36].
Discussion
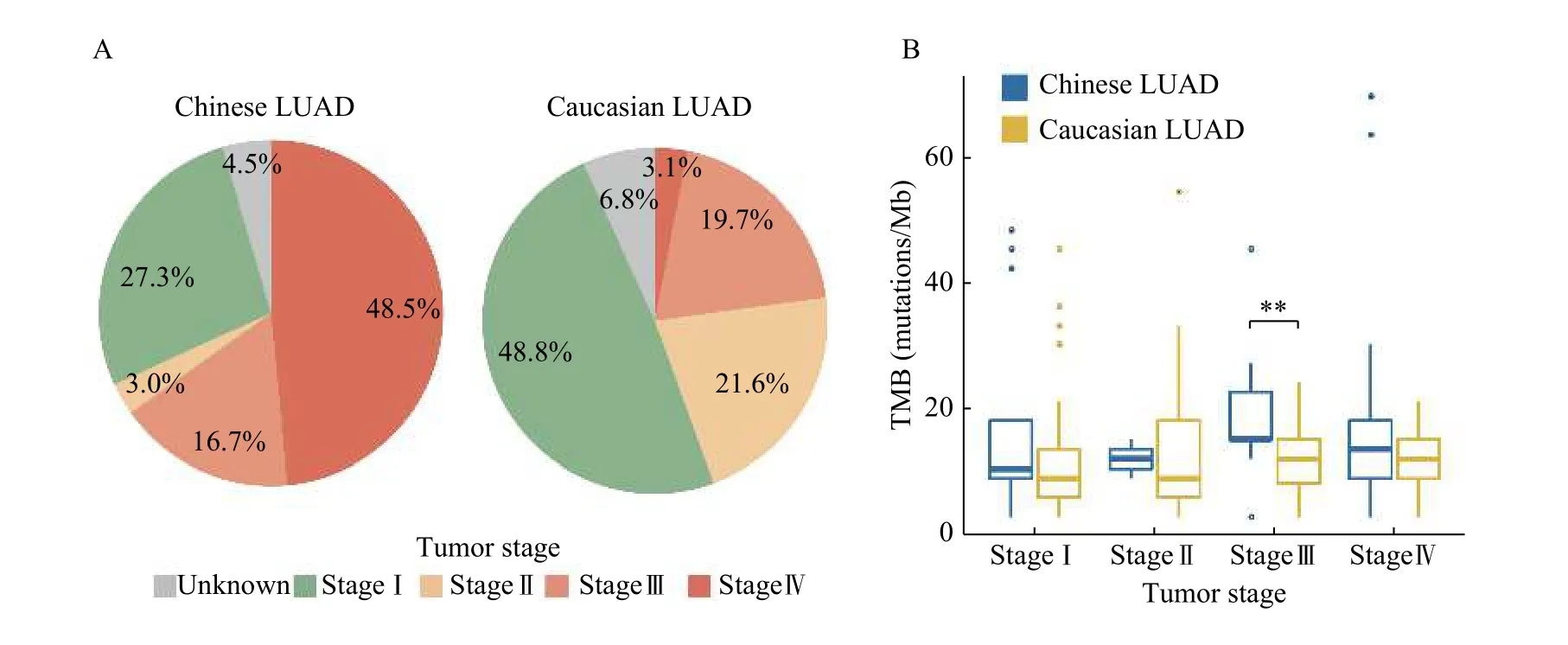
Fig. 4 Tumor mutational burden (TMB) in different tumor stages of LUAD patients. A: Pie charts displaying the tumor stage of LUAD patients. B: Boxplot showing the TMB in different tumor stages. LUAD: lung adenocarcinoma. **P<0.01.
Despite substantial genomic studies on NSCLC have been conducted in European and American populations, sample size targeting Chinese population is relatively small. However, several studies have demonstrated that racial difference does exist in genomic characteristics. To further describe the genetic traits of Chinese patients, we applied targeted sequencing to 66 Chinese LUAD samples, and mutational analysis revealed that missense mutations are common in both cohorts, while C>A transversion events are more frequently detected in Caucasian samples, which is attributed to tobacco smoking.Although tobacco exposure is known as the primary risk factor of lung cancer, LUAD is the most common subtype in Asian female non-smokers. In this study,the proportion of non-smokers in Chinese LUAD is higher than its counterpart (Chinese: 56.1% [37/66];Caucasian: 9.9% [16/162]), and other studies observe the same phenomenon. Previous research revealed that the high incidence of lung cancer in Chinese nonsmokers may be associated with second-hand smoke and cooking fumes[37].
Besides that mutantTP53is frequently detected in both populations, the alteration rates of many genes show racial divergence.EGFRandCREBBPare inclined to alter in Chinese, whereasKRASandKEAP1are inclined to alter in Caucasian samples(Supplementary Fig. 2A). The contrastive mutant rates in driver geneKRASandEGFRindicated that Chinese and Caucasian may have different tumorigenesis mechanisms. The high incidence ofEGFRmutation in Chinese population suggests a benefit from EGFR-TKIs treatment[38]. However, the mutation loci determine the therapeutic efficiency.Exon 19 deletions and L858R mutation in exon 21 are sensitive to EGFR-TKIs, while samples harboring exon 20 insertion or T790M gain resistance to these inhibitors[39-40]. Therefore, precise identification ofEGFRmutation is especially critical to Chinese LUAD patients.
Moreover, we foundEGFRandKRASare driver genes regardless of ethnic communities. Additionally,we identified two novel driver genes,GNASandJAK1, that are specific to the Chinese population.Further, we observed an intensely clustered mutation distribution in Chinese LUAD.
The tumor mutational burden is defined by the number of somatic mutations per megabyte, and lung cancer is known to carry high TMB[41]. Studies have shown that higher TMB is associated with better response to immune checkpoint inhibitors. We discovered that TMB of patients increases with tumor stage, and patients at an advanced-stage harbored higher TMB than at the early stage. At total of 65.2%of patients in our cohort are at advanced stage, so it is important to assess the TMB of LUAD patients before immunotherapy.
Limited by the sample size, we just caught a glimpse of the Chinese LUAD genomics, and did not take the differential sequencing depth between targeted sequencing and WES into consideration. In addition, surgical resection is the first-line treatment for the patients in the early stage, and the patients of advanced stage usually accept targeted therapy after surgery, before which targeted sequencing is implemented. As a result, more advanced patients are included in our study. Additional clinical samples and validation cohorts will be needed to explore the role of novel driver genes of Chinese LUAD, and to further comprehensively decipher the difference between various ethnic groups.
Acknowledgments
This research was supported by grants from projects supported by the National Natural Science Foundation of China (91959113, 81972358, and 81572893), the Natural Science Foundation of Jiangsu Province(BK20180036 and BE2017733).
杂志排行

THE JOURNAL OF BIOMEDICAL RESEARCH的其它文章
- Contained local compression on peri-ascending aortic area for postoperative bleeding control: a case report
- Intrathecal hematoma and sacral radiculitis following repeat epidural blood patch
- Valproic acid therapy decreases serum 25-hydroxyvitamin D level in female infants and toddlers with epilepsy— a pilot longitudinal study
- The testis-specifically expressed gene Trim69 is not essential for fertility in mice
- α-ketoglutarate promotes the specialization of primordial germ cell-like cells through regulating epigenetic reprogramming
- Identification of four novel prognosis biomarkers and potential therapeutic drugs for human colorectal cancer by bioinformatics analysis