First-principles study of the co-effect of carbon doping and oxygen vacancies in ZnO photocatalyst∗
2021-03-11JiaShi史佳LeiWang王蕾andQiangGu顾强
Jia Shi(史佳), Lei Wang(王蕾), and Qiang Gu(顾强)
Department of Physics,University of Science and Technology Beijing,Beijing 100083,China
Keywords: first-principles theory,electron density of states and band structure of crystalline solids,III–V and II–VI semiconductors
1. Introduction
Since 1972 Fujishima and Honda discovered that TiO2electrodes would split water into hydrogen and oxygen regarding photocatalytic effect,[1]semiconductor-based photocatalyst received extensive attention for being the promising material to solve environmental pollution and energy-short issues,including various metal oxide.[2–5]Similar to TiO2, ZnO is an excellent candidate for photochemistry applications due to its high photosensitivity,[6]nontoxicity, and low cost.[7]ZnO possesses the direct band gap and higher photo-generated carriers mobility than TiO2.[8]Photocatalysis in practice deeply depends on efficient light absorption and carriers mobility,but ZnO only can utilize the UV region(~2%of solar energy)of solar energy owing to its large band gap about 3.4 eV.[9,10]Besides, doping single defect into ZnO probably has limitations for efficient carries transfer. Hence,tackling this challenge to find a feasible approach for tuning the energy band structure of ZnO for efficiently utilizing visible light (~45% of solar energy)is an urgent affair.
Very recently, numerous experimental and theoretical studies of non-metal doping in ZnO with better photocatalytic activity have been reported, i.e., N, S, C.[6,11–18]Owing that non-metal elements have closer atom radius to replace O atom, it is much easier to fascinate in experiments than metal incorporated into ZnO lattice, and avoid inducing Shockley junction. The C-doping in ZnO can significantly improve its porosity which demonstrates a larger specific surface applied for higher visible-light respond for photoelectrochemical applications.[11]Nowadays, the research proves that Cdoping ZnO narrows the direct band gap, leading to red shift of light absorption region,meanwhile easily give rise to structural defect appearance.[16]Besides,the experimental investigations emphasis that native defect in ZnO such as Vzn (Zn vacancy) or Vo can evidently suppress the photo-generated carries recombination and improve the visible-light absorption,which offers the excellent enhancement of photocatalytic efficiency.[17]Furthermore,it is worth noting that exceptional catalytic activity is attributed to co-doped the different arrangements for pairs of various dopants which attracted more attention as reported nowadays, such as (Al, Ni) co-doped ZnO,(P,N)co-doped TiO2.[19,20]It has suggested that the coeffect of various defects has compensated effect to remediate the shortcoming caused by the individual dopant within experiments and theoretical work,[4,21]whereby we present discussion towards photocatalytic hydrogen generation regarding the co-effect of C-doping and Vo in ZnO. Accordingly,there are some essential questions existed in catalytic reaction progress the ZnO participated. First,the porosity of C-doping ZnO caused by the occurrence of Vo would result in photoinduced holes and electrons recombination.[11,21]It is crucial to clarify the role of C and Vo respectively if they co-effect in ZnO simultaneously, the origin of light response as well. On the other hand,different concentrations of Vo in visible-lightdriven photocatalyst have a high possibility to induce various effects of the catalytic reaction.[22]Discovering the underlying physical mechanism of the co-effect of C-doping and Vo in ZnO is a fundamental pathway to interpret experimental results, then, it is also important to explore the appropriate amount of Vo in C-doping ZnO for obtaining desired ability upon photocatalytic applications.
In this work, we carry out first-principles calculations to investigate the electronic structure, density of states (DOS),and effective masses of carriers, absorption spectrum of ZnOxC0.0625(x = 0.9375, 0.875, 0.8125). The co-effect of extensive defects in ZnO may improve the photocatalytic performance and provide guidance to devise new optical and optoelectronic materials.
2. Theoretical method and models
Frist-principles calculations as the predictive means of theoretical study for decades, it is widely used to make a connection between experimental observations and theoretical understanding, and further manufacture models for giving clues to predict new desired materials. We perform first-principles calculations using Vienna ab initio simulations package (VASP)[23]based on density functional theory(DFT). The electron–ion interaction of element is described by the projector augment wave(PAW).[24,25]Pseudo-potential selected is the Perdew–Burke–Ernzerof (PBE) exchange–correlation functional within the generalized gradient approximation(GGA).[26]The valance configuration of Zn is 4s23d10,and O is 2s22p4,respectively.
The supercell of 2×2×2 established consists of 32 atoms,which can facilitate the calculations of the co-effect of Cdoping and Vo in ZnO with various conditions, that is,ZnOxC0.0625(x =0.9375, 0.875, 0.8125) systematically analyzed. C atom replaces one O atom formed the C-dopant,the Vo is made by removing an interior O atom from the supercell. Wave functions are expanded by a plane-wave with a cutoff energy of 500 eV and Monkhorst–Pack grids within the Brillouin zone is selected using a 4×4×4 mesh with a structure optimized,then add to a 7×7×7 mesh when we proceed accurate DOS simulations. The lattice constant of wurtzite ZnO with fully relaxed atomic positions are a=3.25 ˚A and c = 5.21 ˚A, which are in agreement with the experimental value.[27,28]The self-consistent convergence to the tolerance is set as 1×10−6eV/˚A,and the maximal force of total energy convergence is set as 0.001 eV/˚A on atoms.
Conventional local density approximation (LDA)[29]or GGA is employed to investigate the energy band of materials contained open-shell electrons, i.e., transition metal oxide, which deviates from experimental evidence. Those approaches only describe well the valence band of ZnO which is strongly correlated material, but underestimate Coulomb interaction of conduction band and overestimate the interplay of the Zn–O bond. In order to solve this deviation, the GGA+U method is used to correct the conduction band by adding the Hubbard-U to restore the real potential between electrons of narrow bands. Comparing with other theoretical beyond DFT scheme, such as Heyd–Scuseria–Ernzerh(HSE)[30]screened hybrid density functional or GW[31]approximation,those methods will spend more time in calculating dozens of atom systems than GGA+U[32,33]and still have an improper description of the band structure. The main problem lies in the underestimation of Coulombic interactions of 3d orbital electrons on Zn. Previous work suggested that the optimum choices are Ud=10.0 eV on Zn and Up=7.0 eV on O which were systemically investigated with geometry optimized or fixed atoms positions.[33]The optical properties of Ce-doping ZnO obtained by the GGA+U using this Hubbard U pairs had proven to work well.[34]In short,the investigation of the electronic structure of the co-effect of C-doping and Vo in Zn photocatalyst within GGA+U method is a fast and effective means of revealing its profound theoretical understanding in our work.
3. Results and discussion
3.1. Structural optimization and formation energy
The crystal structure of ZnO is the hexagonal wurtzite phase with space group p63mc which has thermodynamic stabilities under ambient environment. According to previous work,Vo was always discussed alone by standard DFT or beyond DFT calculations,[25,31–35]rarely concerned co-effect of dopant and Vo on ZnO. Vo as the native defect is easily induced by incorporating heterogeneous elements into ZnO. In C-doping ZnO,experiments observed that the lattice parameter of C-doping ZnO is smaller than that of pure ZnO due to the presence of Vo.[16]Also, Vo plays a role to maintain system neutrality.Hence,we firstly investigate the structure properties of ZnO with the coexistence of C-doping and Vo. To gain the stable crystal structure,we concern that the C-doping with different Vo concentrations affect geometry optimization of ZnO.The supercell was chosen to module the lattice structure of ZnOxC0.0625(x=0.9375, 0.875, 0.8125), the x refers to the percent of oxygen atoms, namely, the 1 −x represents Vo concentrations in system. The possible doping regions are listed below: I.body; II.face,III.edge. The body region has four possible sites that can be substituted by C atom due to the crystal symmetry of the hexagonal phase(see Fig.1(a)),illustrated that the formation energies of those doping configurations are basically same. The stable structure of C-doped ZnO contained Vo is obtained by the optimization of the supercell of ZnO0.875C0.0625(remove one oxygen atom from supercell)and ZnO0.8125C0.0625(remove two oxygen atoms from supercell) are shown in Figs. 1(b) and 1(c). The oxygen atom is removed one by one after C atom located in a stable position.For the case of Zn0.875C0.0625, forming five possible configurations: (i) C-doping + body atom II, (ii) C-doping + body atom III,(iii)C-doping+body atom IV,(iv)C-doping+face atom, (v) C-doping + edge atom. It is found that the system energy of ground states favors(i)and(ii)configurations whose energies are the same as each other. In this paper,the research of Zn0.875C0.0625is based on the(ii)configurations. The crystal structure which contains the higher Vo concentrations will have four possible doping positions, which are marked as: i)C-doping+Vo+body atom II,ii)C-doping+Vo+body atom IV,iii)C-doping+Vo+face atom,iv)C-doping+Vo+edge atom. The iv) configuration is chosen as the stable configuration for Zn0.8125C0.0625due to its lowest energy. The lattice constants relaxed with different doping configurations are listed in Table 1,showing that the crystal date is in good agreement with the experimental measurements.16,33,34In the case of Zn0.9375C0.0625,C anion has a greater radius than O,therefore, it is reasonable to infer that the co-effect of C-doping and Vo in ZnO has a smaller lattice constant and cell volume because of the occurrence of Vo.
In the following works, we focus on the co-effect of Cdoping and Vo in ZnO based on this lattice structure. The crystal structure of ZnO0.875C0.0625optimized is shown in Fig.2. C atom is the center of the tetrahedron whose vertexes composed of four Zn atoms. Substitution of O by C bonded with adjacent Zn atom yields length about 1.94 ˚A,which is shorter than 1.98 ˚A of the original O–Zn bond. Zn atoms from the basal plane of tetrahedron move forward to C atom, thereby, the geometry structure is affected by the enhanced interaction between Zn and C. Besides, the existence of Vo leads to the chemical internal pressure changed and slightly squeeze C atom towards the basal plane. There is volumetric shrinkage and stronger bonding interactions than both pure Zn and ZnO0.8125C0.0625. The higher Vo concentrations in ZnO0.8125C0.0625will influence the stable crystal symmetry due to the more Zn–O bonds broken, the crystal structure prefers to the deformation which further impacts the electronic structure. More importantly,Vo will improve the porosity that is reasonably attributed to inter-particle slits.It has a great possibility that the co-effect of C-doping and Vo provides more free carries to benefit photochemical applications.
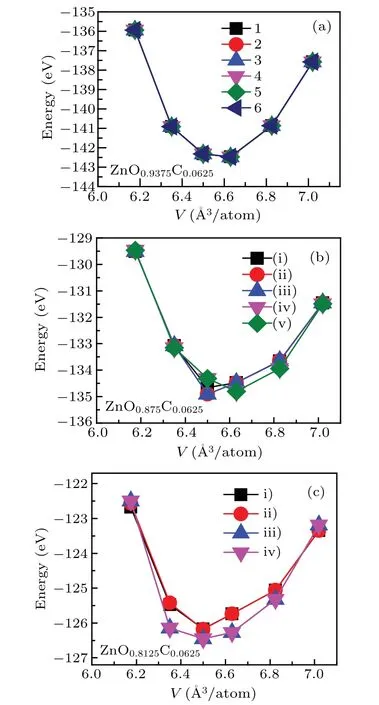
Fig.1. The energies as a function of volume for various doping configurations in(a)ZnO0.9375C0.0625,(b)ZnO0.875C0.0625,and(c)ZnO0.8125C0.0625.

Table 1. The comparison between the calculation results and experimental results regarding the ZnOxC0.0625 (x=0.9375,0.875,0.8125)on both lattice parameters and energy band gap.
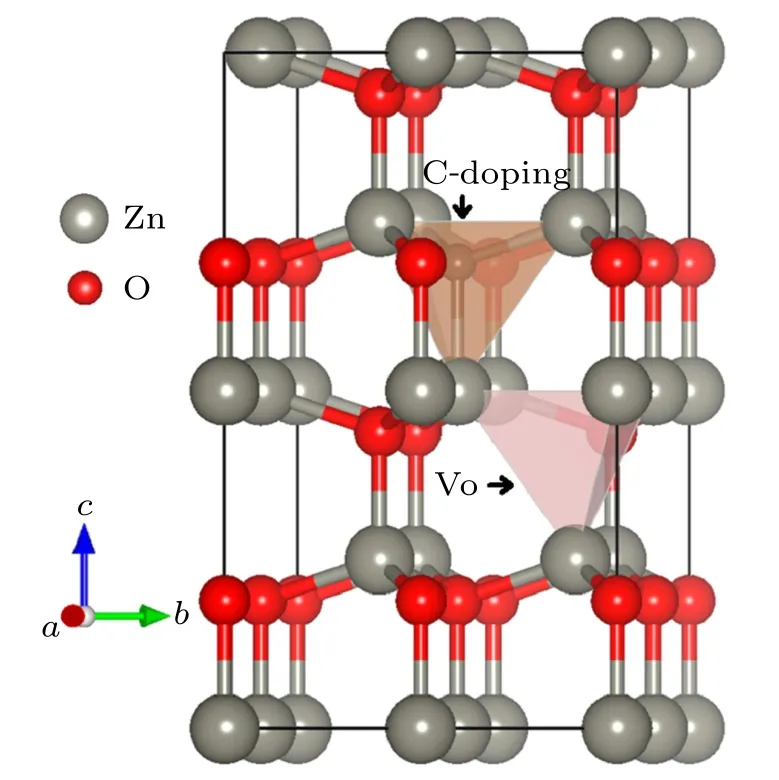
Fig.2. Optimized structure of ZnO0.875C0.0625,illustrating that co-effect of C-doping(one oxygen atom replaced by carbon atom)and Vo(remove one oxygen atom from supercell)in ZnO.
3.2. Electronic properties
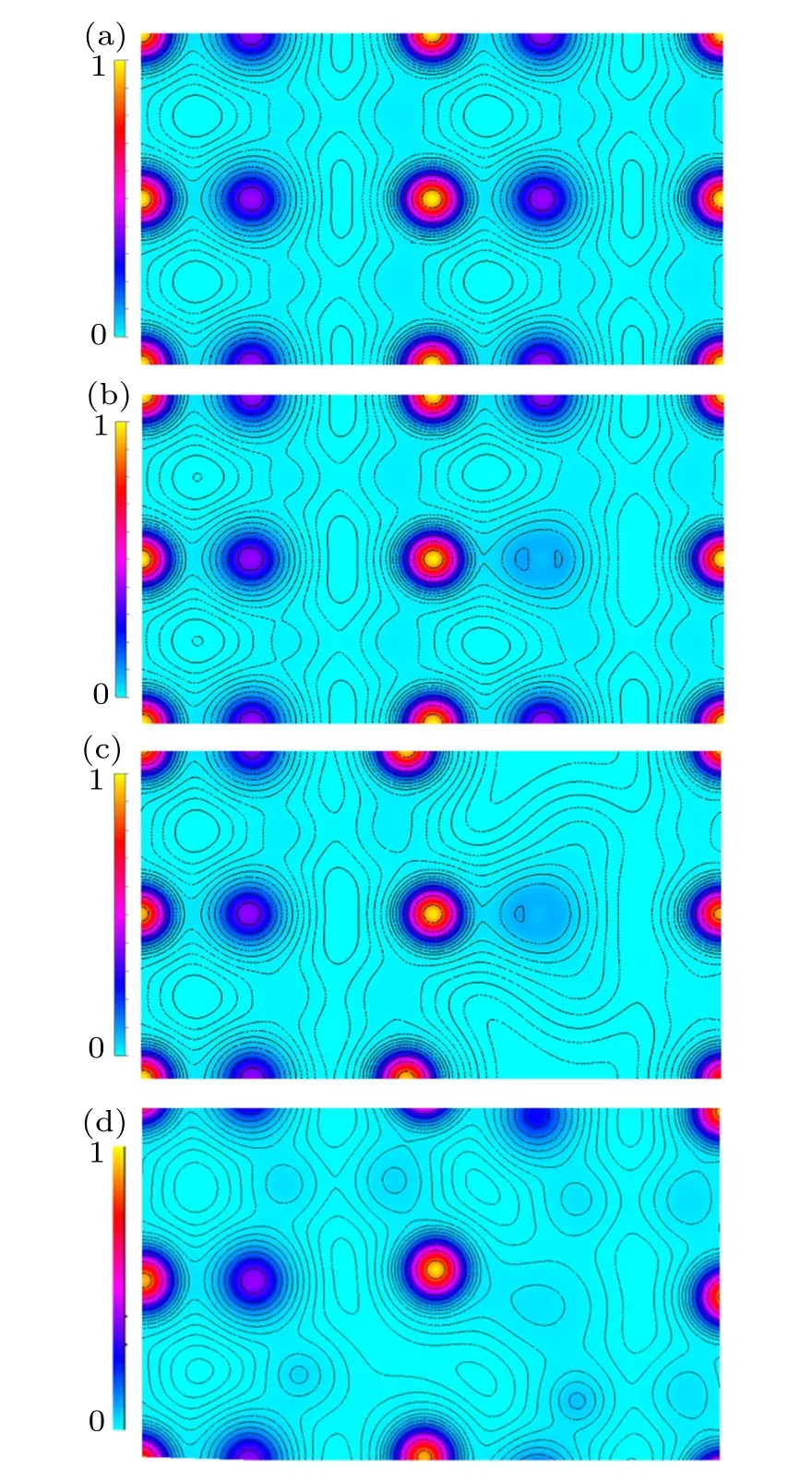
Fig.3.The calculated valence–electron density on the(100)plane in(a)pure ZnO,(b)ZnO0.9375C0.0625,(c)ZnO0.875C0.0625,and(d)ZnO0.8125C0.0625.
Taking the underlying understanding of the co-effect of C-doping and Vo in ZnO into consideration, here we need to clarify the role of C-doping and Vo separately. The charge densities of ZnO,ZnOxC0.0625(x=0.9375,0.875,0.8125)are shown in Fig.3. The picture of Fig.3(a)illustrated that pure ZnO has a neat charge density distribution calculated on the(100)plane. Each of Zn atom is formed a stable bond with the nearest O atom. By comparison, the non-equilibrium charge distribution at C–Zn is depicted in Fig.3(b) which refers to ZnO0.9375C0.0625, C bonded with the nearest Zn forming π–π bond has a stronger interplay than Zn–O bond, meanwhile excluding the chance to form chemical bonds with others Zn.Furthermore, four Zn–O bonds are broken immediately once one Vo appeared,the charge is redistributed to form the chemical bonding states in ZnO0.875C0.0625as shown in Fig.3(c).The dangling bonds remain partially filled by two electrons and hence can accommodate six more. This simple chemical picture dictates Vo acts as a deep donor. Unfilled C-2p orbital direct connects with Zn-3d electrons. We present the charge redistribution in Fig.3(d)concerned the absence of two oxygen atoms. The enlarged defect region makes more dangling bonds between C and Zn. It is supposed to have more trapping sites to capture itinerant electrons or holes resulting in the negative effect on the catalysis efficiency.Although the C–Zn bond possesses stronger covalence nature than Zn bonding with O,Vo dose as a donor for the system charge distribution equilibrium. And that volumetric shrinkage with high porosity gains lager specific surface in ZnO on the account of Vo emerged.
Photocatalysis properties are typically associated with the band structure and density of states (DOS). Herein, the band structure and DOS of ZnOxC0.0625(x=0.9375, 0.875,0.8125)are systemically investigated. Based on the GGA+U band structure of pure ZnO,ZnOxC0.0625(x=0.9375, 0.875,0.8125) are shown in Figs. 4(a), 4(b), 4(c), and 4(d), respectively. The band gap of pure ZnO calculated by standard DFT scheme is about 0.78 eV as the Zn-3d orbital located in the higher energy states.[16,30,31]Adopting the U value correction, the energy band gap of ZnO enlarges to about 2.2 eV.The calculated band gap is also listed in Table 1. From the fold Brillouin zone of a supercell, this makes it more reasonable to lead to computational results in small differences with 3.4 eV of experimental observations.[33,36]The direct band gap at Gamma point in the Brillouin zone determines the scope of solar energy and facilitates that photoexcited carries from valence band transit to conduction band. In the case of ZnO0.9375C0.0625,the band gap can be essentially narrowed without changing curvatures of band structure,suggesting that the C dopant expands absorption range to visible light spectrum without affecting the mobility of carriers. According to previous work, Vo as the donor can release electrons to narrow the band gap and reconstruct the shape of the band edge,for improving the possibility of carries which reach the surface of a semiconductor to participate in chemical reaction.[34]Considering the co-effect of C-doping and Vo in ZnO,valence band edge shifts up while the conduction band edge moves down, leading to evident reduction of bandgap with curve almost unchanged in ZnO0.875C0.0625. There is indicated that the absorption spectrum will red shift towards visible region concerned the effect of various defects on the electronic structure. Meanwhile, even though the higher Vo proportion of ZnO brings about the smaller energy band gap, the curves of band structure nearing by Fermi level tend to be flat, and the charges near the Fermi level tend to be localized as illustrated in Fig.4(d)in terms of ZnO0.8125C0.0625.Therefore,it is worth noting that the existence of Vo can contribute electrons to the system for improving photocatalytic activity. Theoretical investigations suggest that ZnO0.875C0.0625has a higher visible light response for photocatalytic applications than other two compositions.
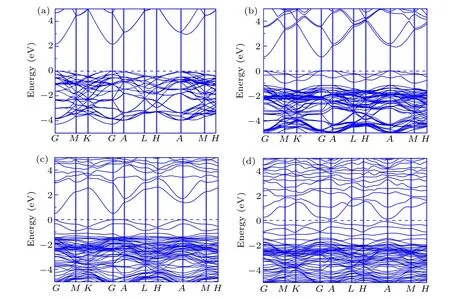
Fig.4.Band structure of(a)pure ZnO,(b)ZnO0.9375C0.0625,(c)ZnO0.875C0.0625,(d)ZnO0.8125C0.0625 within the frame of GGA+U calculation,respectively. The Fermi level is set as 0 eV.
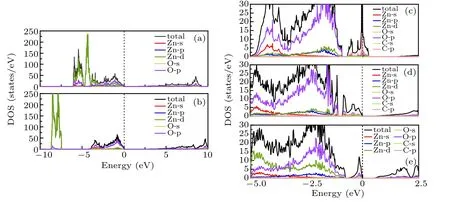
Fig.5. The electronic DOS of(a)pure ZnO is obtained by GGA method. The DOS of(b)pure ZnO,(c)ZnO0.9375C0.0625,(d)ZnO0.875C0.0625,(e)ZnO0.8125C0.0625 obtained by GGA+U method.
To figure out the influence of orbital electrons on the electronic structure of ZnOxC0.0625(x=0.9375, 0.875, 0.8125)in ZnO, the total density of states (TDOS) and partial density states (PDOS) are calculated separately and showed in Fig.5. Owing to GGA calculations underestimate interactions between transition metal 3d or 4f electrons, from Fig.5(a), it is found that Zn-3d states are located at about −6.5 eV.Zn-3d states are corrected to about −8.2 eV (experimental average value ~8.27 eV[33]) shown in Fig.5(b). The DOS of pure ZnO indicates that the valence band edge mainly consists of O-2p states and Zn-3d states along with some contributions from O-2s states,while the conduction band composes of Zn-3d and Zn-4s states. The DOS of Fig.5(c) presents that the valence band edge of ZnO0.9375C0.0625is mostly occupied by C-2p electrons. This observation affirms that C has significant contributions for the reduction of band gap instead of the strong hybridization of O and Zn orbitals. In the case of the ZnO0.875C0.0625, we identify DOS featured as having valence band dominated by C-2p, O-2p, at the same time, given the contributions from the Zn-3d. The conduction band is mainly attributed by Zn-4s electrons. The conduction band localized nearby the Fermi level is suggested that Vo as a donor plays the role of charge center in the n-type semiconductor. The weaker Zn–O covalent bond can be easily broken, which is reflected by the “pseudogap” of DOS. A higher DOS of O-2p states emerge in ZnO0.8125C0.0625, which originates from a larger number of broken bonds between Zn and O.Thereby,more carriers easily trapped by Vo leading to catalysis reacting passivation are generated.
3.3. Carriers effective masses and absorption spectrum
The effective masses of carriers are determined by the curvatures of valence bands and conduction bands. The mobility of electrons and holes photo-excited is inversely proportional to effective masses, which is closely related to the amounts of carriers involved chemical reactions for affecting photocatalytic activity. Photogenerated carriers usually undergo a redox reaction on the surface of a semiconductor,but it is also highly possible to cause electrons and holes recombination, thereby, reducing the redox reaction rate. There is speculation that Vo may be the recombination center for harvesting photogenerated carries. Besides,different dopant levels have different influences on the mobility of visible light drive carriers. Therefore, the effective masses of charges at first are essentially investigated in the terms of ZnOxC0.0625(x=0.9375, 0.875, 0.8125). The effective masses can fairly estimate charge carriers mobility fitting by parabolic portions of the CBM (conduction band minimum) and VBM (valence band maximum).[24]We evaluate the effective masses of electrons and holes along three directions in the Brillouin zone using the following equation:
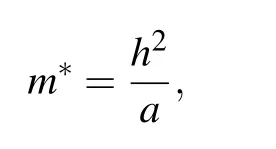

Table 2. The ratio value of effective masses between electronsand holes of ZnOxC0.0625 (x=0.9375, 0.875, 0.8125) and ZnOx(x=0.9375, 0.875) obtained from the edge of band gap by parabolic fitting along three directions in the Brillouin zone.

Table 2. The ratio value of effective masses between electronsand holes of ZnOxC0.0625 (x=0.9375, 0.875, 0.8125) and ZnOx(x=0.9375, 0.875) obtained from the edge of band gap by parabolic fitting along three directions in the Brillouin zone.
Direction [010] [001] [210]m∗e/m∗h m∗e/m∗h m∗e/m∗h ZnO0.8125C0.0625 0.48 1.02 0.58 ZnO0.875C0.0625 0.83 0.29 0.20 ZnO0.9375C0.0625 0.18 0.0009 0.48 ZnO 0.05 0.01 0.37 ZnO0.9375 0.13 7.44 0.21 ZnO0.875 0.06 0.008 0.07
A further discussion we focused on the absorption spectrum, which manifests the relatively straightforward application of light absorption and is expected to clarify the role of C-doping and Vo separately upon visible-light-responsive catalysis. The results of the absorption spectrum are shown in Fig.6. The limitation of visible light absorption of ZnO originates from the wide band gap. The highlighted part of ZnO refers to energy from 1 eV to 2.8 eV, which is the most favorable range for photocatalysis. The ZnO0.9375C0.0625associated with this light behavior tends to be natural. Remarkably, ZnO0.8125C0.0625utilizes more photoluminescence than ZnO0.875C0.0625at the beginning of the favorable part, nevertheless, the absorb spectroscopy reverse to the latter. In the view of experiments, the most measuring band gap is around at 3.0 eV by controlling the dopants or vacancies.[16,33,34]That means the optical characterization of ZnO0.8125C0.0625is below our expectations. This is due to large Vo concentrations have more dangling bonds which would catch holes to enhance p–d hybrid and electrons localized, which is also confirmed by the results of effective masses ratio. ZnO0.8125C0.0625and ZnO0.875C0.0625have slightly different macroscopic phenomena under the visible light respond region. We concern the electronic structure and charge density on equal footing, the ZnO0.875C0.0625has moderate carries mobility, most importantly, it possesses the most prepared light absorption scope and larger specific surface originated from the porous structure. Herein, we believe that ZnO0.875C0.0625is the most promising candidate for improving the catalysis under illumination within our work.
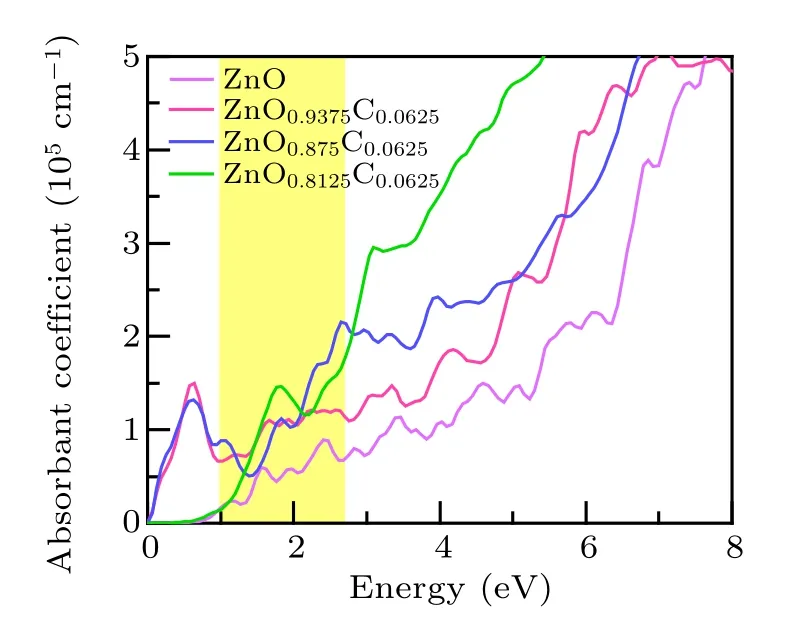
Fig.6. The light absorption spectra of pure ZnO,ZnOxC0.0625 (x=0.9375,0.875, 0.8125)are obtained by GGA+U method, respectively. The yellow part refers to the region in which ZnO-based materials are easy to absorb.
4. Conclusions
The present studies reveal that the co-effect of Cdoping and Vo indeed have enhanced photocatalytic performance in ZnO by calculating the electronic properties of the ZnOxC0.0625(x=0.9375,0.875,0.8125). The method we carried out is the GGA+U formula based on density functional theory. It is indicated that the optimal system of co-existence is the ZnO0.875C0.0625, which has a controllable band gap in the visible light region with an improved absorbed application, and lower ratio value of effective masses of carries to avoid recombination between holes and electrons. Such electronic behavior driven by the illumination is of benefit to have the expected photocatalysis activity for generating hydrogen.Also, we found that Vo in ZnOxC0.0625(x=0.9375, 0.875,0.8125) acts as a donor to equilibrate localized charge distribution and gives aid to bond C with Zn. Generally,C-doping domains the valence band lifted to have the narrower band gap in ZnO,and the improved visible-light absorption is mainly attributed to the presence of Vo with light concentrations simultaneously. We predicted the optimal configurations of the coexistence of both C-doping and Vo with more effective on photocatalytic application, but also elucidated the distinguished roles between C-doping and Vo upon the collectively effects on photocatalyst ZnO.
猜你喜欢
杂志排行

Chinese Physics B的其它文章
- Statistical potentials for 3D structure evaluation:From proteins to RNAs∗
- Identification of denatured and normal biological tissues based on compressed sensing and refined composite multi-scale fuzzy entropy during high intensity focused ultrasound treatment∗
- Folding nucleus and unfolding dynamics of protein 2GB1∗
- Quantitative coherence analysis of dual phase grating x-ray interferometry with source grating∗
- An electromagnetic view of relay time in propagation of neural signals∗
- Negative photoconductivity in low-dimensional materials∗