NOTCH3基因新发位点突变致伴有皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病1例报告
2021-03-09史兆博马丽丽
史兆博, 孙 永, 马丽丽
伴有皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病(cerebral autosomal dominant arteriopathy with subcorticaliInfarcts and leukoencephalopathy,CADASIL)是一种由NOTCH3基因突变引起的进行性加重的脑小动脉病[1]。基因检测发现Notch3基因特征性的半胱氨酸改变、活体组织检查发现嗜锇颗粒沉积或免疫组织化学显示Notch3蛋白染色呈阳性,可明确诊断CADASIL[2]。本例患者基因测序发现NOCTH3基因6号外显子杂合错义突变,氨基酸改变为p.Y337C(第337号氨基酸由酪氨酸变异为半胱氨酸),人类基因突变数据库专业版(human gene mutation database professional edition,HGMD pro)已报道与CADASIL相关,核苷酸改变为c.1010A>G(编码区第1010号核苷酸由腺嘌呤变异为鸟嘌呤),HGMD pro未发现相同位点突变,现报道如下。
1 病例资料
患者,男性,61岁,农民,小学文化,自2019年5月开始逐渐出现行走时双下肢发沉,行走速度减慢,发作性视物成双,具体虚实相位不能描述,每次持续数分钟缓解,每天发作数次,应用活血化瘀药物10 d后视物成双症状未再发作,规律服用阿司匹林肠溶片100 mg/晚;阿托伐他汀钙片10 mg/晚,逐渐出现记忆力下降并进行性加重,近记忆力下降为主,双下肢发沉症状持续不缓解,步基增宽,经常出现小便失禁;2020年6月开始出现视物模糊,双眼右视时明显,具体性质不能描述,无眼前发黑感,反复发作,每次持续数分钟,起初每3~5天发作一次,逐渐发展至每天发作约5次,遂于2020年8月2日于“开封市中心医院”住院诊疗。既往11 y前“脑梗死”,表现为右侧肢体力弱,无明显后遗症;30多岁时有反复偏头痛发作史,40岁以后很少发作;近4 y性格改变,脾气暴躁,执拗;无“高血压病、糖尿病、高脂血症、冠心病”病史;否认吸烟史,偶尔少量饮酒(平均2~4次/y,饮酒量<2两/次)。患者父亲及1兄分别在60岁、53岁时因“脑梗死”去世。入院时体温、脉搏、呼吸、血压及内科系统查体未见明显异常;神经系统查体:记忆力及远记忆力均有减退、Romberg征睁闭眼均不稳;宽基底步态、行走缓慢,四肢肌力、肌张力正常,双侧病理征阴性。头部磁共振血管成像未见明显异常;脑组织磁共振平扫及弥散加权成像(Diffusion Weighted Imaging,DWI)(见图1);脑组织磁敏感功能成像(Susceptibility Weighted Imaging,SWI)(见图2);根据患者脑组织磁共振结果,结合CADASIL筛查量表[3]评分18分,考虑诊断为高度可疑CADASIL,送检基因测序,结果(见图3),停用“阿司匹林肠溶片”,给予“西洛他唑胶囊,早晚各100 mg口服”,同时给予活血化瘀、改善循环、保护神经元线粒体功能及他汀类药物应用,治疗1 w后患者症状未再发作,遂出院。
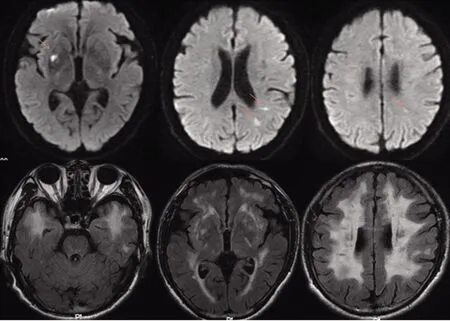
图1 DWI提示右侧基底节、左侧枕叶多发急性脑梗死;FLAIR提示双侧弥漫性脑白质病变
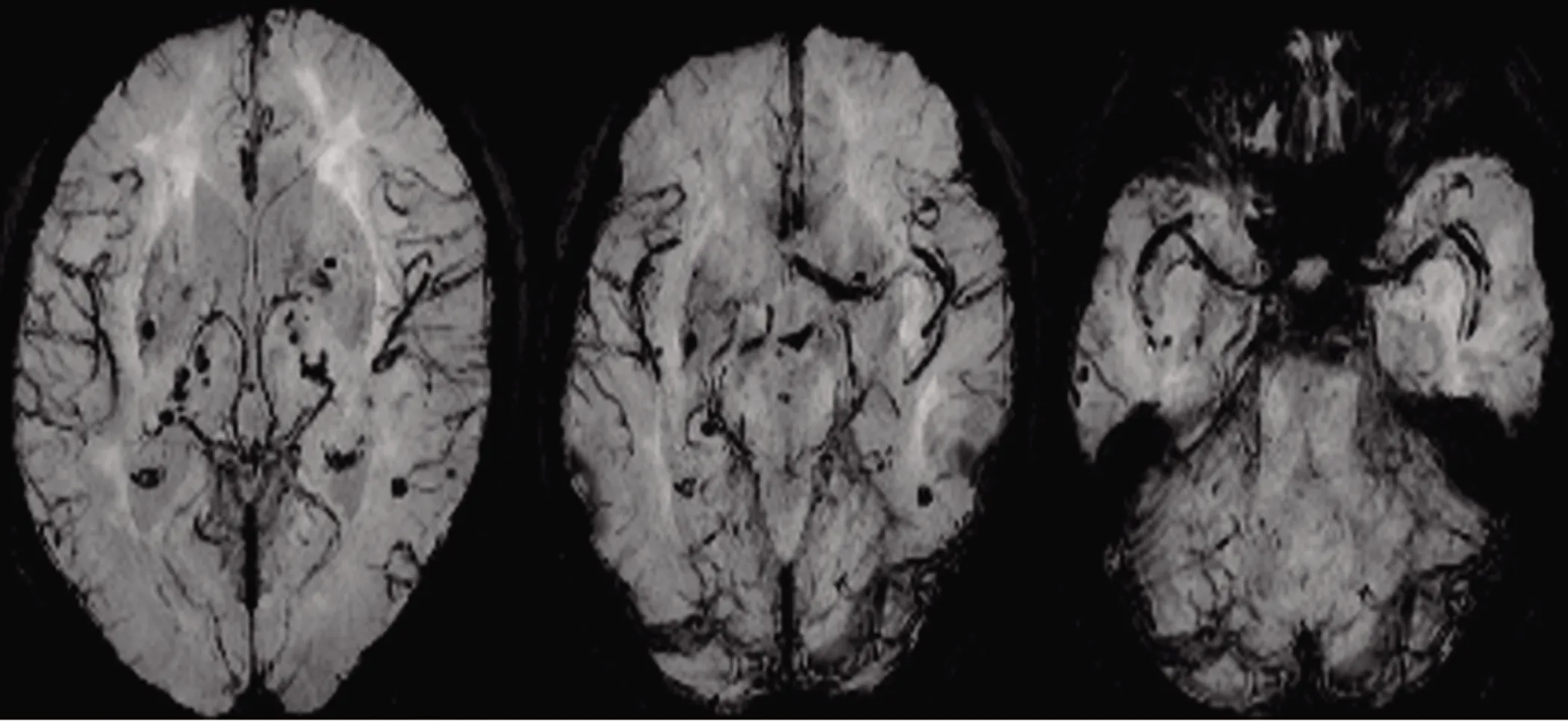
图2 SWI提示双侧基底节区,颞枕叶皮质下多发含铁血黄素沉积
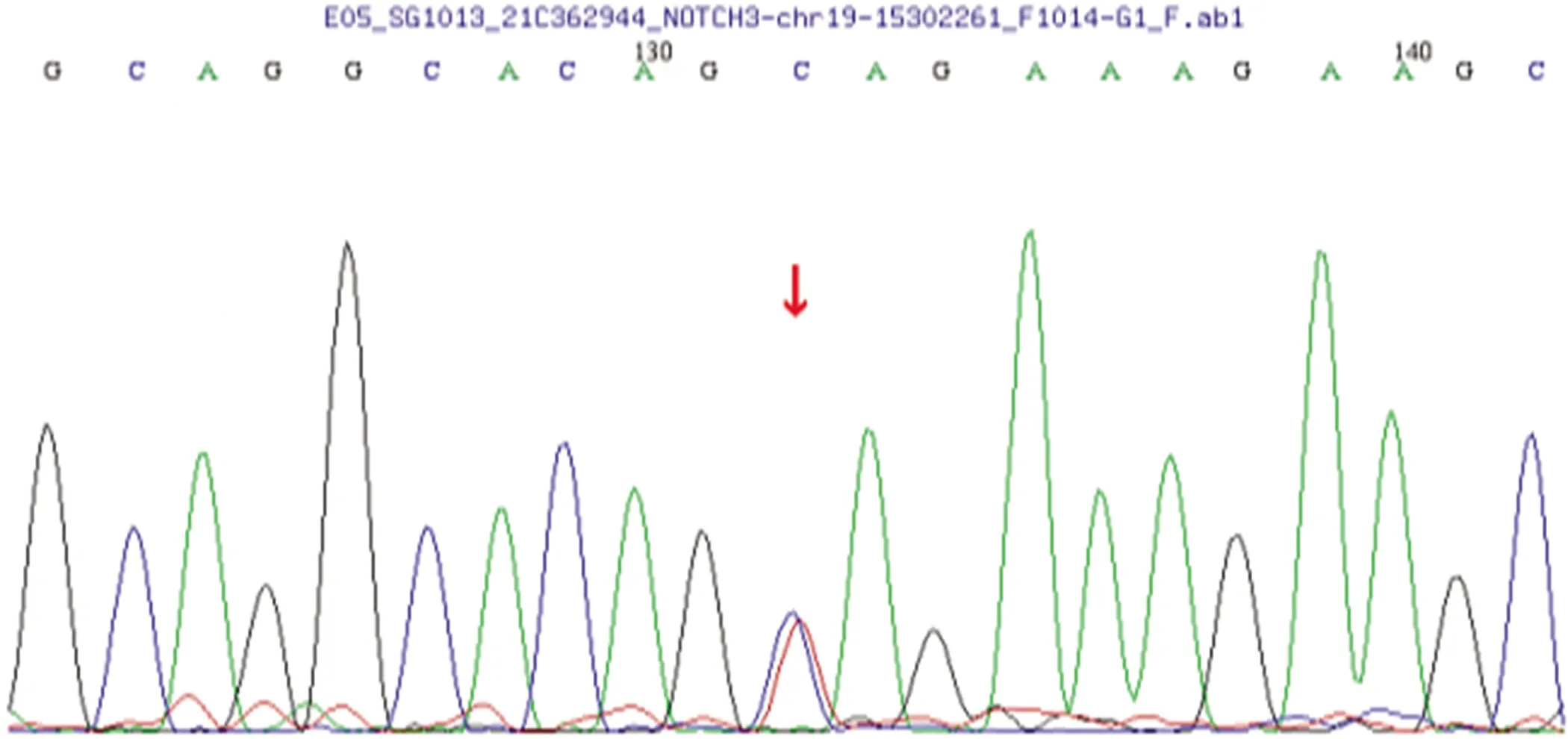
图3 Notch3基因第6号外显子区域发现一处杂合突变点:c.1010 A>G导致氨基酸改变p.Y337C;HGMD pro数据库未见报道
2 讨 论
CADASIL是由Notch3基因突变引起的一种最常见的遗传性小血管病,一般中年起病,常无高血压、糖尿病、高脂血症、吸烟等常见的脑血管病危险因素,以反复发作的脑卒中为主要临床表现,伴随精神障碍、情感淡漠、步态障碍、认知功能下降并逐步进展为痴呆,约半数患者青年期有先兆性偏头痛[4~6]。NOTCH3基因位于染色体19p13.1-13.2区域,CADASIL相关的基因突变分布在34个EGFr中,导致了这些EGFr中半胱氨酸残基的增加或减少,破坏了二硫键的原有结构,使其编码的跨膜蛋白失发生结构或功能的变化,变异的NOTCH3蛋白聚集体(颗粒状嗜锇物质)沉积在小动脉血管壁的平滑肌细胞表面,包括脑、肾脏、脾脏、心肌、肌肉、皮肤的小动脉和毛细血管等[7],累及脑小动脉从而引起皮质下梗死和弥漫性脑白质高信号(White Matter Hyperintensities,WMH),脑组织SWI可见脑深部白质含铁血黄素沉积[8]等典型头部磁共振成像(MRI)表现,其中颞叶或外囊的脑白质病变对该病的诊断有特征性意义[9]。
HMGD pro数据库可以检索到458种NOTCH3基因突变,大部分为错义/无义突变;该患者外周血样本检测到NOTCH3基因6号外显子杂合错义突变,氨基酸改变为p.Y337C,HGMDpro 数据库已报道与CADASIL相关,Opherk已报道的p.Y337C所对应的碱基改变为c.1088A>G(编码区第1088号核苷酸由腺嘌呤变异为鸟嘌呤),但尚未经功能验证确定为致病性突变(PP5)[10],该患者核苷酸改变为c.1010A>G,查阅既往文献及HGMD pro,未发现相同位点突变,但未经父母样本验证(PM6)。查询突变在正常人群基因库中的突变频率,包括人类外显子数据库、千人基因组突变频率数据库、人群基因组突变频率数据库,在正常人群中无携带率(PM2);经过Mutation Taster(0.999)、SIFT(0.014)、Provean(-5.38)、Polyphen-2(0.999)等预测软件分析,结果均提示有害(PP3),且其SIFT、Provean及Polyphen-2预测的有害性明显高于c.1088A>G(预测值分别为1.0,1.14,0.045)。
根据美国遗传学与基因组学学会(the American College of Medical Genetics and Genomics,ACMG)指南,致病性分析为可能致病性突变,证据为:PM2+PM6+PP3+PP5。
本例患者反复发作脑卒中,伴随性格改变、步态障碍及认知功能下降,青年期有偏头痛病史,符合CADASIL的临床特征;除此之外,患者还有一项突出的症状,即小便失禁,这在脑小血管病中较为常见[11]。国内报道的1例NOTCH3基因11号外显子c.1630 C>T (p.R5447C)突变患者(76岁),除典型CADASIL临床特征外,伴随小便失禁[12]。Dichgan等总结了来自于29个病理确诊的家系中的102例CADASIL患者(30~66岁)的表型谱,其中有28%患有痴呆,在痴呆患者中,86%患有小便失禁[13]。Opherk等对411例CADASIL患者随访观察了10 y,80%患者出现了小便失禁[10];LADIS研究认为,重度WMH为尿急、尿失禁的独立危险因素[14]。该患者的突出影像学表现为弥漫性脑白质脱髓鞘,可能为其尿失禁的原因之一;此外,该患者WMH累及双侧颞极及外囊,DWI可见多发皮质下腔隙性脑梗死,SWI可见多发微出血,多集中在基底节区,颞枕叶皮质下也可见微出血病灶,符合CADASIL的典型影像学特征。
本例提示,对于无明显危险因素的反复发作性脑梗死合并双侧对称脑白质病变的患者,应详细追问偏头痛病史及家族史并考虑到遗传性脑小血管病的可能性,结合CADASIL量表筛查,有助于指定初步诊疗方案,进一步基因筛查有助于明确诊断。本例NOTCH3基因新发位点突变丰富了基因突变数据库,但仍需完善该家系其他成员的基因筛查,未来将进一步追踪随访以期建立完善的家系图谱。
