磷添加方式对NiMo/Al2O3催化剂加氢脱硫性能的影响
2021-03-08汪佩华秦志峰吴琼笑李聪明苗茂谦常丽萍孙鹏程曾剑王立华谢克昌
汪佩华,秦志峰,,3,吴琼笑,李聪明,苗茂谦,常丽萍,孙鹏程,曾剑,王立华,谢克昌
(1 太原理工大学煤科学与技术教育部与山西省重点实验室,山西太原030024;2 山西省环境规划院博士后科研工作站,山西太原030002;3 清华大学环境科学与工程博士后流动站,北京100084;4 山西道生鑫宇清洁能源有限公司,山西岢岚036300)
焦炉煤气常被用作燃料和化工原料,其作为化工原料深加工(合成甲醇、合成氨、合成天然气、合成油、合成乙二醇等)经济效益最好[1-6]。焦炉煤气中除主要成分H2、CH4、CO 等外,还有H2S、CS2、COS、RSH、C4H4S 等不同形态的硫化物,其存在会造成深加工过程各类催化剂中毒失活,如焦炉煤气合成天然气使用的Ni 系催化剂,其吸硫0.15%(质量分数)时活性下降50%,吸硫0.5%(质量分数)活性基本丧失,且失活催化剂均不可再生[7],所以焦炉煤气合成化学品前必须进行深度脱硫。随着焦炉煤气深加工规模的大型化,原料气中总硫脱除精度要求从0.15mg/m3降至0.03mg/m3[8]。在所有硫化物中噻吩结构最稳定,且对催化剂毒性最强[9],如何对噻吩进行深度脱除是焦炉煤气深加工面临的主要难题。
焦炉煤气深度脱硫过程一般采用二级加氢脱硫(HDS)工艺[10-11],在其净化工艺中加氢催化剂的性能至关重要,受到研究者广泛关注。随着原料气中总硫脱除精度提高,传统的HDS 催化剂,如NiMo/Al2O3、CoMo/Al2O3、FeMo/Al2O3等对噻吩的HDS 性能需要进一步提高,以满足日益严格的深度净化要求。研究表明,添加助剂改性传统HDS 催化剂是提高其HDS 性能的重要途径之一。研究发现,在加氢催化剂中引入磷助剂可以明显改善催化剂的结构和物化性质[12],例如在Ni(Co)Mo/Al2O3中引入P 后,降低了活性金属组分Mo 与载体间的相互作用,提高了Mo 在催化剂表面的分散[13],增强了催化剂表面酸性,进而提高了HDS 活性[14];比较MoCoNi/Al2O3引入P 前后发现,引入P 后催化剂表面八面体配位的Mo 物种增多,四配位Mo 物种降低,更有利于高活性Ⅱ型Mo-Co-Ni-S 相的生成[15]。此外,适量P 的加入能够提高催化剂的活性,过量则导致其活性下降,其原因是催化剂表面形成了相对稳定的Co(Ni)-Mo-P 物种,使活性组分Mo 难以还原[16],当催化剂P助剂的负载质量为1%时活性达到最大。
传统加氢脱硫催化剂制备一般采用共浸渍的方式[17-20],即先配制好含P、Ni、Mo 的前体浸渍液,然后浸渍于Al2O3载体上,通过干燥和焙烧获得所需催化剂。研究表明,磷的添加方式对催化剂的物化性质产生重要的影响,从而影响催化剂的加氢脱硫性能。有文献报道,磷添加方式会影响煤焦油加氢脱芳性能,先浸渍P 和Ni 后再浸渍Mo 的浸渍顺序能促进催化剂表面活性金属组分分散均匀,加氢脱芳率达到67.18%[12]。胡乃方等[21]研究发现先浸渍P,然后再浸渍Mo,最后再浸渍Co,这样多步浸渍方式得到的催化剂HDS 活性最佳,转化率达96.98%。由此可知,不同制备方式制备的改性催化剂对于煤焦油中不同物质的脱除程度不一样,虽然焦炉煤气与煤焦油组成和性质差异较大,但其改性方法为脱硫催化剂的设计提供了很好的理论指导。目前所知,采用不同磷添加方式制备P改性催化剂对焦炉煤气中噻吩HDS 性能的深入研究,有可能实现同种催化剂在不同领域的转化应用,进一步阐明磷添加方式对NiMo/Al2O3催化剂应用于焦炉煤气中噻吩HDS性能的作用机理。
本文制备了5种不同磷(1%P,质量分数)添加方式改性的系列NiMo/Al2O3催化剂,并对其应用于焦炉煤气中噻吩的HDS 活性进行了评价研究。通过BET、X 射线衍射(XRD)、H2程序升温还原(H2-TPR)、NH3程序升温脱附(NH3-TPD)、C4H4S(H2)程序升温脱附[C4H4S(H2)-TPD]、X射线光电子能谱(XPS)、高清透射电镜(HRTEM) 和拉曼(Raman)等分析手段对催化剂的物化性质进行了表征,系统探究了不同磷添加方式对催化剂结构及HDS活性相的影响。
1 实验部分
1.1 实验药品
所有药品均购自药品供应商并未经过纯化处理:γ-Al2O3,淄博贝格工贸有限公司;七钼酸铵,天津市化学试剂四厂,AR,(NH4)6Mo7O24·4H2O质量分数≥99.0%;硝酸镍,上海沃凯生物技术有限公司,AR,Ni(NO3)2·6H2O质量分数≥98.0%;磷酸铵,天津市光复精细化工研究所,AR,(NH4)3PO4·3H2O质量分数≥98.5%;柠檬酸,天津市科密欧化学试剂有限公司,AR,C6H8O7·H2O质量分数≥99.5%。
1.2 催化剂的制备
1.2.1 NiMo/Al催化剂的制备
催化剂采用等体积浸渍法制备,其制备过程如下:γ-Al2O3载体使用前在120℃下干燥5h,并测量其吸水率;将一定量的七钼酸铵和柠檬酸溶于去离子水中搅拌至完全溶解,再加入硝酸镍制成浸渍液,加入γ-Al2O3浸渍得到包含5% NiO 和15% MoO3的催化剂前体,100℃下干燥5h,500℃下煅烧3h,得到NiMo/γ-Al2O3催化剂,催化剂标记为NiMo/Al。
1.2.2 P助剂改性NiMo/Al催化剂的制备
催化剂采用分步浸渍法制备。第一种方式是先用磷酸铵配制成浸渍液,加入γ-Al2O3载体浸渍,100℃下干燥5h,500℃焙烧3h 后获得改性载体;然后使用柠檬酸配制七钼酸铵和硝酸镍的稳定混合溶液,改性载体浸渍后获得前体,100℃下干燥5h,500℃下煅烧3h,得到P-NiMo/γ-Al2O3催化剂,催化剂标记为P-NiMo/Al。
第二种方式是先用柠檬酸、磷酸铵和硝酸镍配制成浸渍液,加入γ-Al2O3载体浸渍,100℃下干燥5h,500℃下煅烧3h,得到含1% P 和5% NiO 的γ-Al2O3前体,标记为PNi/Al;然后使用七钼酸铵配制成含Mo 浸渍液,将PNi/Al 前体浸渍,100℃下干燥5h,500℃下煅烧3h,得到P 改性的PNi-Mo/Al催化剂。
第三种方式是先用柠檬酸、磷酸铵和七钼酸铵配制成浸渍液,加入γ-Al2O3载体浸渍,100℃下干燥5h,500℃下煅烧3h,得到含1%P 和15%MoO3的γ-Al2O3前体,标记为PMo/Al;然后使用硝酸镍配制成含Ni浸渍液,将PMo/Al前体浸渍,100℃下干燥5h,500℃下煅烧3h,得到P改性的PMo-Ni/Al催化剂。
第四种方式是先用七钼酸铵和柠檬酸溶于去离子水中搅拌至完全溶解,再加入硝酸镍制成浸渍液,加入γ-Al2O3载体浸渍,100℃下干燥5h,500℃下煅烧3h,得到含5% NiO 和15% MoO3的γ-Al2O3前体,标记为NiMo/Al,然后使用磷酸铵配制成含P浸渍液,将NiMo/Al前体浸渍,100℃下干燥5h,500℃下煅烧3h,得到P改性的NiMo-P/Al催化剂。
第五种方式是直接在硝酸镍和七钼酸铵配制成的浸渍液中引入磷酸铵,加入γ-Al2O3载体浸渍,100℃下干燥5h,500℃下煅烧3h,得到含1%P、5%NiO和15%MoO3的NiMoP/Al催化剂样品。
1.3 催化剂表征
采用北京金埃谱科技有限公司生产的VSorb4800 型物理吸附仪(BET)在-196℃条件下对制备的催化剂进行N2物理吸脱附分析。样品先在真空中300℃下脱气6h,冷却至室温后转移至吸附仪上进行后续吸附。利用多点BET方法从吸附-脱附等温线计算催化剂比表面积和孔体积,通过BJH法测定样品的孔径和孔径分布。
使用日本理学Rigaku MiniFlex600 型X 射线衍射仪(XRD)对催化剂表面物相进行定性分析,采用Cu Kα射线(λ=1.54178Å,1Å=0.1nm),管电压40kV,管电流15mA,扫描范围为5°~85°,扫描步长0.02°,扫描速度8°/min。通过与JCPDS 卡片对照确定物相的构成。
使用浙江泛泰仪器有限公司FINSORB-3010型化学吸附仪对催化剂进行H2-TPR 测试来分析催化剂表面金属还原性。将100mg 40~60目的催化剂样品放置于U 形石英反应管中,Ar 气氛下升温到300℃预处理30min 除去表面吸附杂质,然后Ar 吹扫冷却到室温,切换10%H2-90%Ar混合气进行还原反应,气体流量为20mL/min,待基线平稳后,以10℃/min 的升温速率从室温升到900℃,然后Ar吹扫降温。
使用浙江泛泰仪器有限公司FINSORB-3010型化学吸附仪对催化剂进行NH3-TPD 和C4H4S(H2)-TPD 分析。NH3-TPD 测试过程如下:将100mg 催化剂样品(40~60 目)放置于石英U 形反应管中,在He 气氛中300℃下预处理30min,然后He 吹扫冷却至室温,通入3% NH3/Ar 混合气以20mL/min 的流量吸附30min 后,继续吹扫60min 至基线平稳,然后从室温以10℃/min的升温速率升温至800℃进行脱附,完成后He 吹扫降温。C4H4S(H2)-TPD分析:将100mg催化剂样品(40~60目)放置于石英U 形反应管中,通入H2和1.5%H2S/N2混合气于400℃下进行原位预硫化60min,然后He 气氛下吹扫冷却至室温,通入0.1%C4H4S/N2(10%H2-90%Ar)混合气以20mL/min 的流量常温吸附30min后,He吹扫60min至基线信号平稳,然后从室温以10℃/min 的升温速率升温至500℃进行脱附,脱附完成后He吹扫降温。
硫化态催化剂表面元素状态由Thermal VG 公司生产的Escalab Xi+型X 射线光电子能谱分析仪(XPS)进行测试,采用单色Al Kα射线源,将粉末状催化剂样品装入双面胶带上放入真空室进行分析,分析条件下的真空度为2×10-7Pa。在分析过程中采用外来污染碳的C1s作为基准峰对样品进行荷电校正,以测量值和参考值(284.8eV)之差作为荷电校正值(Δ)来校正谱图中其他元素结合能。
采用InVia-Reflex 型激光拉曼光谱仪(Raman) 对样品表面分子结构进行拉曼分析(520nm 的He-Ne 激光),探测器扫描范围100~1500cm-1,步长1cm-1。入射光采用发射线照射,使6mW 的光束通过系统显微镜聚焦于样品,样品的拉曼光谱在室温下使用配有二级管阵列检测器的拉曼微探针进行检测。
催化剂表面形貌及粒径分布在JEOL JEM-2010F型高分辨透射电子显微镜(HRTEM)上进行。测试前先将少量经过钝化的样品放入无水乙醇中,超声60min,使待测样品尽可能分散,然后用洁净的滴管取样置于覆有碳膜的300目铜网上,经红外干燥后将样品以氮气流的形式转移到显微镜的样品架上。
1.4 催化剂活性测试
催化剂样品在微型固定床反应装置上进行加氢脱硫(HDS)性能评价,气氛采用模拟焦炉煤气,其组分如表1所示(为了便于测试,气氛中噻吩高于真实气氛)。称取40~60 目500mg 催化剂样品放于反应器恒温区(床层高径比=3∶1),催化剂床层上下装有惰性陶瓷管支撑。新鲜加氢催化剂均是以氧化态形式存在,而氧化态催化剂的加氢活性、稳定性、选择性、抗毒性及寿命等均低于硫化态催化剂,因此加氢催化剂使用前需进行预硫化处理,催化剂预硫化是在流速为20mL/min H2气氛下升温至180℃,通入1.5% H2S/N2混合气并保持30min,继续升温到400℃预硫化60min;切换N2吹扫床层降温至250℃,再切换原料气稳定反应2h后开始取样测试。采用北京中科惠分仪器有限公司GC-7820双FPD 检测器气相色谱仪分析,检测条件为:氮气(99.999%)作为载气、流量3.3mL/min、FPD检测器温度为260℃、检测器高压700V、分离柱为HP-Sulfur 型毛细管柱[尺寸为50m×0.53mm×7.0μm(膜厚)],测量过程柱箱温度采用程序升温,以15℃/min 从0℃升温至90℃进行测试,以使不同形态硫易于分离且色谱峰分布均匀,FPD检测器的最低检出限为0.75mg/m3。反应压力1.0MPa、反应温度250~390℃(从250℃开始采样,之后每隔20℃稳定反应30min 进行数据采集),空速为4000h-1。为了提高空速调整噻吩进口浓度,测试过程中向模拟焦炉煤气中配入1/2惰性气体N2,测试过程噻吩进口浓度稀释为292.5mg/m3。HDS过程中噻吩转化为硫化氢和羰基硫,因此反应前后S 平衡(nS)及噻吩转化率(XC4H4S,%)可按式(1)和式(2)计算。

表1 模拟焦炉煤气气氛组成

式中,nH2S为出口硫化氢物质的量,mol;nCOS为出口羰基硫物质的量,mol;nC4H4S,out为出口噻吩物质的量,mol;Cin和Cout分别为反应气在床层进口和出口中C4H4S浓度,mg/m3。
2 结果与讨论
2.1 催化剂HDS性能评价
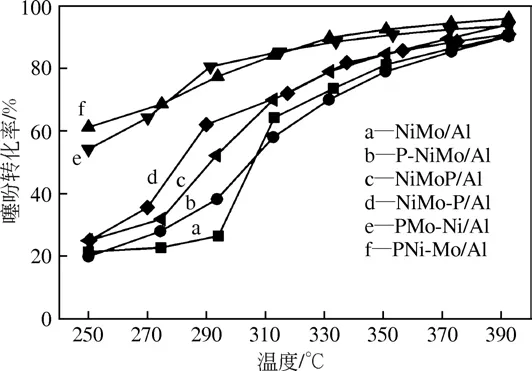
图1 不同催化剂的HDS活性
图1为不同磷添加方式制备系列催化剂在焦炉煤气气氛下对噻吩的HDS 活性评价。可以看出,噻吩的HDS 转化率随温度升高逐渐增大,当温度升高至390℃时,HDS 转化率接近,均大于90%,不同磷添加方式制备催化剂对噻吩的低温HDS 活性影响较大。当反应温度T≤270℃时,PNi-Mo/Al和PMo-Ni/Al两种磷添加方式制备催化剂对噻吩的HDS 活性远大于其他催化剂,其在250℃下噻吩HDS 转化率分别达61%和54%。研究认为噻吩的脱硫有两种路径[22-23]:一种是通过在环状结构上直接加氢生成四氢噻吩,然后其上C—S 键断裂,最终脱去环中的S 原子,称为加氢路径(HYD);第二种是其上C—S 键直接断裂生成丁二烯,然后再通过加氢反应得到丁烯以及丁烷,称为直接脱硫路径(DDS)。文献中认为,DDS途径消耗的氢较少,是一种较好的路径,且更容易在低温下进行,当加入1%P后DDS反应速率增加了90%[16,24]。推测为不同磷添加方式制备催化剂改变了DDS 反应速率,因此在低温度下活性表现出较大差别。
2.2 催化剂的BET表征
表2 为Al2O3载体及不同磷添加方式制备系列催化剂的BET 分析结果。与γ-Al2O3载体比较,负载后催化剂比表面积和孔体积均减小,这是由于活性组分在载体孔道沉积造成的。P/Al、PNi/Al、PMo/Al 分别为P-NiMo/Al、PNi-Mo/Al、PMo-Ni/Al这3种催化剂分步浸渍过程的中间体,其与NiMoP/Al 共浸渍制备催化剂比表面积接近,而经过分步浸渍制备的P-NiMo/Al、PNi-Mo/Al、PMo-Ni/Al 催化剂比表面积和孔体积则下降较多,其原因是经过分步浸渍制备催化剂过程二次煅烧对催化剂的孔道结构造成破坏。P-NiMo/Al、PNi-Mo/Al、PMo-Ni/Al 催化剂的孔径增大,可能是先浸渍磷对载体起到一定的扩孔作用或者经过负载活性组分后载体表面微孔数量减小,导致平均孔径增大。
2.3 XRD分析
图2(a)为不同磷添加方式制备系列催化剂硫化前的XRD谱图。催化剂在2θ为37°、46°、67°的衍射峰归属于Al2O3的特征峰(JCPDS No. 10-0425)[25],同时谱图中没有检测到MoO3和NiO的特征峰,说明活性组分分散性较好。同NiMo/Al2O3催化剂相比,引入P 后的催化剂谱图出现了NiMoO4的特征峰,在2θ 为26.7°处为β-NiMoO4的特征峰(JCPDS No.45-0142),2θ=14.2°、25.4°、28.7°、32.5°处 为α-NiMoO4的 特 征 峰(JCPDS No.33-0948)[26-27],NiMoO4相是催化剂表面分散状态的Ni2+与Mo 相互作用形成的,对催化剂HDS 反应具有积极作用,能够降低催化剂表面MoO3的还原温度,促进加氢脱硫Ⅱ型活性相Ni-Mo-S 的形成[28]。不同磷添加方式制备催化剂表面形成的β-NiMoO4晶相有差异,其中,PNi-Mo/Al、PMo-Ni/Al 和P-NiMo/Al 这3 种催化剂表面β-NiMoO4峰较强且窄,说明其上形成了较多β-NiMoO4晶相。
研究认为,Al2O3表面有两种晶格空位,分别为晶格四面体空位和晶格八面体空位,对于负载于Al2O3表面的金属氧化物,焙烧过程中其优先进入Al2O3表面晶格四面体空位,形成与载体相互作用较强的MAl2O4物种,其次再进入表面晶格的八面体空位,形成与载体作用相对较弱的物质[29]。因此,推测PNi-Mo/Al、PMo-Ni/Al 和P-NiMo/Al 这3种催化剂制备过程P 优先进入Al2O3表面晶格四面体空位中形成了AlPO4物种,阻止Ni 和Mo 进入Al2O3表面晶格四面体空位,而进入Al2O3表面晶格八面体空位,形成与载体相互作用较弱的物种,促进其形成NiMoO4相。

表2 催化剂物理性能
先浸渍磷制备P-NiMo/Al催化剂过程中,因为P与Al2O3之间强的亲和力导致金属与载体之间的相互作用力过低,活性金属颗粒发生聚集,降低了活性金属的分散度,这也是P-NiMo/Al催化剂表面生成β-NiMoO4相多但活性较低的主要原因。图2(b)是硫化后的催化剂XRD 谱图,各催化剂表面除了Al2O3的特征峰外,还有2θ 为33.6°和59.3°处的MoS2特征峰(JCPDS No.17-0744),而且PNi-Mo/Al、PMo-Ni/Al 两种磷添加方式制备的催化剂表面活性物种MoS2衍射峰较强,说明这两种磷添加方式更容易在催化剂表面形成HDS 活性物种MoS2,因此具有更高的HDS活性。

图2 催化剂硫化前后的XRD谱图
2.4 H2-TPR分析
图3 为不同磷添加方式制备系列催化剂的H2-TPR 谱图。NiMo/Al 催化剂在407℃的还原峰是八面体配位Mo 物种中Mo6+到Mo4+的还原[30-31],762℃处的还原峰是Mo4+和四面体配位Mo 物种中Mo6+到Mo 的还原[32]。同NiMo/Al 催化剂比较,通过不同方式引入P 后,在300~600℃间出现2~3 个新的还原峰,催化剂NiMoP/Al、NiMo-P/Al 第一个还原峰为Mo6+的还原,第二个还原峰则为Ni、Mo 相互作用α-NiMoO4的还原;PNi-Mo/Al、PMo-Ni/Al和P-NiMo/Al 催化剂除了上述两个峰外,第3 个还原峰为β-NiMoO4的还原。由于P 与载体优先作用减弱了金属组分NiMo 与载体间的相互作用,可以看出762℃处的还原峰变小甚至消失,说明P 的加入减少了四面体配位难还原物种的数量。先浸渍磷制备的P-NiMo/Al 催化剂的低温还原峰向右偏移,这是由于活性金属颗粒发生聚集导致金属分散度下降难还原所致,该现象与XRD 及催化剂评价结果相一致。同时研究认为,NiMoO4的还原分为两步,第一步是在较低温度下还原成NiMo 合金,还有部分MoO2,第二步是高温下MoO2的进一步还原,MoO2在催化剂还原硫化过程中容易形成HDS 活性物种MoS2[33]。因此,在不同磷添加方式制备的催化剂中,PNi-Mo/Al、PMo-Ni/Al 催化剂促进了Ni 和Mo 在载体表面分散,生成更多的有效NiMoO4相,促进了HDS活性物种生成。

图3 催化剂H2-TPR谱图
2.5 NH3-TPD分析
不同磷添加方式制备系列催化剂的NH3-TPD谱图如图4所示。所有催化剂的NH3-TPD曲线在130℃、400℃和610℃处左右均有3个脱附峰,分别对应弱(<300℃)、中(300~550℃)、强(>550℃)酸性位点[17]。载体Al2O3表面主要以弱酸和中强酸为主,引入P 后,载体表面酸性发生明显变化,可以看出,改性载体P/Al 表面弱酸量增加,中强酸消失;负载活性金属组分NiMo 后,各催化剂上主要以弱酸位为主,包括少量的中强酸和强酸位。不同方式引入P 后,催化剂弱酸位发生明显偏移,催化剂PNi-Mo/Al 的NH3解吸峰温度最低,P-NiMo/Al 最高,两者温差接近50℃,催化剂PNi-Mo/Al表面弱酸位浓度较高,强酸位浓度较低,表明P的引入方式改变了催化剂表面酸性。研究认为,金属氧化物在载体表面的分散性与载体的酸性密切相关,酸性位点浓度越高,越有利于金属原子的沉积,催化剂PNi-Mo/Al 表面高浓度的弱酸位为活性金属组分沉积提供了有利空间,从而提高了Mo 在载体表面的分散,形成了较多易还原硫化的Mo 物种[34-35],这也是PNi-Mo/Al催化剂HDS活性较高的原因。

图4 催化剂NH3-TPD谱图
2.6 C4H4S(H2)-TPD分析
图5 为不同硫化态催化剂表面C4H4S-TPD 和H2-TPD 谱图。从图中看出,催化剂脱附曲线主要有弱吸附解离峰Ⅰ和强吸附解离峰Ⅱ,对应反应物C4H4S和H2的两种吸附活性位。文献报道,硫化态催化剂上噻吩HDS反应活性位是S空位(NiMoS团簇边缘配位不饱和的金属中心),分为Mo边S空位和S 边S 空位,而这些空位能够吸附反应物分子[36-37],说明在催化剂表面存在不同的噻吩吸附位,这也是硫化态催化剂出现两种不同脱附曲线的主要原因。从图5(a)和(b)中可以看出,催化剂表面强吸附活性位较弱,吸附活性位数量多且吸附结合能大,强吸附活性位吸附结合能大导致其解离脱附能大,反应物分子(C4H4S 和H2)解离脱附难度增加,而在弱吸附活性位反应物分子(C4H4S 和H2)容易解离脱附。从C4H4S-TPD 和H2-TPD 谱图可以看出,PNi-Mo/Al 和PMo-Ni/Al 催化剂表面弱吸附解离活性位较多,结合催化剂HDS性能评价结果,表明催化剂表面存在两种加氢活性位,且弱吸附解离活性位对催化剂的性能提高具有重要作用。
2.7 XPS分析
图6(a)为不同磷添加方式制备系列催化剂硫化前表面Mo3d的XPS谱图。从图中看出,NiMo/Al催化剂在电子结合能232.5eV 和235.7eV 处为含有Mo6+物种的峰,可能归属于MoO3或NiMoO4[38-41],同时引入P 后各催化剂表面同样仅有Mo6+物种的峰,说明P的引入没有改变催化剂表面的Mo物种结构。但是,P引入方式的不同导致催化剂的电子结合能相对于NiMo/Al均发生了0.1~0.2eV的偏移,说明P引入方式的不同造成活性金属组分之间相互作用力发生变化。

图5 硫化态催化剂C4H4S-TPD和H2-TPD谱图
图6(b)为不同磷添加方式制备系列催化剂硫化后表面Mo3d 的XPS 谱图。相对于硫化前催化剂表面Mo6+的谱峰,硫化后的催化剂在相应位置的谱峰消失,峰位向低结合能方向发生了偏移,说明在硫化过程中金属组分间产生了相互作用。除了Mo6+的谱峰外,结合能229.8eV 和232.1eV 处为Mo4+物种的峰[42],该峰归属于完全硫化的活性物种MoS2[31];229.7eV 处的峰归属于Mo5+,对应于未被完全硫化的MoOxSy。从图图6(b)中看出,PNi-Mo/Al、PMo-Ni/Al 和NiMo-P/Al 催化剂硫化后Mo4+物种的谱峰面积要大于其他催化剂的谱峰面积(谱峰面积代表表面物种的含量),即催化剂表面生成了更多的活性物种MoS2。尽管NiMo 催化剂活性位点的性质不明确,但Ni-Mo-S 结构是目前被广泛认同的活性位点[43-44],而Ni-Mo-S 结构是活性物质MoS2边缘活性中心Mo与Ni 结合形成的[45],因而MoS2含量越多,说明催化剂上具有更多可以供Ni 结合的边缘活性位点,因此MoS2的数量决定了催化剂的HDS性能。
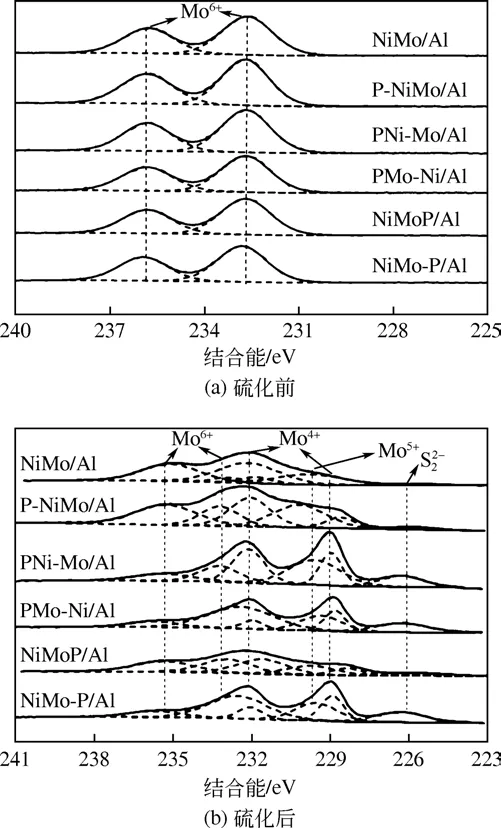
图6 催化剂硫化前后表面Mo3d的XPS谱图
表3 列出了硫化后催化剂中Mo 物种在催化剂表面组成及Ni/(Ni+Mo)、S/Mo 原子比。硫化度用n(Mo4+)/n(Mo)表示,载体与活性金属间相互作用力强弱决定了其在预硫化阶段被硫化程度的大小。由表3可以看出,不同磷引入方式表面硫化度的大小为PNi-Mo/Al>PMo-Ni/Al>NiMo-P/Al>NiMoP/Al>P-NiMo/Al>NiMo/Al,硫化度越大,说明催化剂表面载体与金属组分Mo物种间的相互作用越弱,Mo物种易被硫化,形成的硫化物MoS2较多,HDS 活性位点也就越多。同时表中Ni/(Ni+Mo)原子比能够反应催化剂表面Ni的分散性[46],比值越大,说明催化剂表面Ni 分散性越好,有利于与活性物种MoS2边缘活性位结合;S/Mo 比值越大,说明催化剂表面S 原子浓度越高,Mo 物种的硫化越完全,因而HDS 活性越高,可以看出,PNi-Mo/Al、PMo-Ni/Al催化剂表面Ni/(Ni+Mo)、S/Mo比值最大,这与硫化度结果相一致。
2.8 HRTEM分析
图7为不同P添加方式制备系列催化剂硫化后HRTEM 谱图。可以看出,催化剂表面分布有明显的晶格条纹,其中,晶格间距(d=0.61nm)较大的条纹为层状结构的MoS2晶粒[47],晶格间距(d=0.22nm)较小的条纹为Ni 的硫化物NixSy晶粒,由图7可知,不同磷添加方式制备催化剂的硫化态表面晶格条纹形貌之间存在差异。从图7(a)~(b)中看出,催化剂表面活性金属组分MoS2晶粒分散且数量少,而从图7(c)~(f)可以看出,添加P后硫化态催化剂表面晶格条纹清晰可见,说明催化剂表面形成了较多的MoS2物种;此外,从图7(c)~(d)中可以看出,催化剂PNi-Mo/Al、PMo-Ni/Al 催化剂表面MoS2晶格边缘位存在明显Ni的硫化物晶格,而图7(e)~(f)中 仅 发 现MoS2晶 格 条 纹,说 明PNi-Mo/Al、PMo-Ni/Al催化剂中Ni更容易与MoS2边缘位结合形成具有较高HDS 的Ⅱ类Ni-Mo-S 活性相,因而脱硫活性更高。
2.9 Raman分析
图8为不同磷添加方式制备系列催化剂在100~1300cm-1范围内的Raman谱图。所有催化剂在波数为750~1050cm-1范围内的峰归属于NiMoO4物种的特征峰[48-49],相对NiMo-P/Al 和NiMoP/Al 催化剂在960cm-1处 的 峰,P-NiMo/Al、PNi-Mo/Al 和PMo-Ni/Al 催 化 剂 的 峰 在948cm-1、950cm-1、956cm-1处都发生了偏移,同时其3个催化剂的谱峰发生了宽化,说明样品表面物种与载体的相互作用减弱,活性组分分散性增强;NiMo-P/Al和NiMoP/Al催化剂在830cm-1处出现了晶相MoO3谱带,这一结果与H2-TPR和XRD结果相一致。在波数为213~224cm-1范围内的峰归属于八面体配位的Mo—O—Mo的伸缩振动,主要是α-NiMoO4物种;波数350~363cm-1范围内主要是MoO3的特征谱带[50-51],各催化剂谱峰均有不同程度的偏移,峰强也有不同程度的差异,NiMo-P/Al和NiMoP/Al的峰较强,说明活性金属组分Mo在载体表面的分散性较差。

表3 硫化物催化剂表面Mo物种组成和Ni/(Ni+Mo)、S/Mo原子比

图7 硫化后催化剂HRTEM谱图

图8 催化剂的Raman谱图
3 结论
磷添加方式对NiMo/Al2O3催化剂的HDS性能产生明显影响,PNi-Mo/Al 和PMo-Ni/Al 两种磷添加方式制备催化剂对噻吩的低温HDS 活性远大于其他催化剂,以含292.5mg/m3噻吩的模拟焦炉煤气为原料时,其在250℃下噻吩HDS 转化率分别达61%和54%。通过BET、XRD、H2-TPR、NH3-TPD、C4H4S(H2)-TPD、XPS、HRTEM 和Raman 等手段对催化剂进行了表征,发现PNi-Mo/Al 和PMo-Ni/Al催化剂低温HDS 活性高是因为这两种磷添加方式减弱了活性金属组分NiMo 与载体的相互作用,提高了NiMo 在载体表面的均匀分散,生成能够促进催化剂硫化形成Ⅱ型活性相Ni-Mo-S 的NiMoO4物种,NiMoO4和MoO3之间的协同作用提高了催化剂的硫化度,导致其表面弱吸附解离活性位增多,对反应物分子(C4H4S 和H2)的吸附解离能力增加,使其HDS活性增强。
