稠环芳烃加氢饱和催化剂研究进展
2021-03-08刘道诚王九占荆洁颖杨志奋冯杰李文英
刘道诚,王九占,荆洁颖,杨志奋,冯杰,李文英
(太原理工大学煤科学与技术省部共建国家重点实验室培育基地,山西太原030024)
随着世界经济的发展和人们环保意识的增强,我国对燃油品质的要求越来越高,对提高能源的利用效率日益重视。为提升燃油品质,国Ⅵ标准进一步限制了汽柴油中的芳烃含量(规定汽油中芳烃体积分数≤35%、柴油中稠环芳烃质量分数≤4%),从而降低CO、颗粒物、有毒物的排放,减少发动机燃烧室沉积物的产生[1-2]。而在能源利用效率方面,我国煤焦油的产能和产量随着钢铁产业的发展逐年增加[3]。将煤焦油中的稠环芳烃加氢提质转化为环烷烃,可提升其燃烧性能,用于生产高附加值的燃料油及调和组分,这既能补充石油产品的不足,又能提高煤焦油的附加值,具有良好的发展前景[3-6]。因此对稠环芳烃加氢饱和反应以及加氢饱和催化剂的研究是近年来的研究热点之一。
稠环芳烃的加氢饱和反应已经被学者广泛研究,热力学研究表明,该反应在热力学上可行,且氢压越高、温度越低时加氢平衡转化率越高[7];动力学研究表明,在稠环芳烃逐环加氢饱和过程中其加氢速率常数逐渐减小,想要彻底去除芳香性得到环烷烃变得愈发困难,末环(即加氢饱和至分子中只含一个芳环时)的加氢饱和反应往往是难点所在[8-9]。
分析其原因,主要包括以下三点:①受共振效应的影响,在稠环芳烃加氢饱和至末环的过程中,该不饱和环的共振能逐渐增加,最终接近苯环的共振能,加氢饱和难度增加;②末环不仅共振能较大,还可能被周围的饱和环包裹,空间位阻增大,不利于其在催化剂表面的吸附活化;③在稠环芳烃逐环加氢饱和过程中,所含芳环个数不同的产物之间存在竞争吸附,使得末环的加氢饱和反应受其他中间产物的抑制作用[8,10]。可见,在稠环芳烃的加氢饱和过程中,共振能的变化、空间位阻的存在导致末环在催化剂上吸附困难,同时各产物间的竞争吸附又会抑制末环的加氢饱和反应。
针对上述难点,本文结合稠环芳烃中萘、菲、蒽、芘为主要模型化合物的相关研究,分析了稠环芳烃加氢饱和反应特点和四类稠环芳烃加氢饱和催化剂“构效关系”。总结指出,增加催化剂活性位点的数量和形成活性金属的缺电子状态,能够提高催化剂的加氢活性,促进末环的加氢饱和,并对稠环芳烃加氢饱和催化剂的改进方法提出建议。
1 稠环芳烃加氢饱和反应特点
稠环芳烃的加氢饱和反应为可逆放热反应,为了实现深度加氢饱和,首先要确保反应在热力学上可行,并结合动力学选择适宜的操作条件。侯朝鹏等[11]利用HSC-Chemistry 4.0对蒽和菲加氢反应热力学进行研究,结果表明,压力为5MPa、温度低于350℃时菲的平衡转化率接近100%,产物主要为全氢菲的同分异构体。此外,反应压力会影响产物的平衡组成,通过提高压力能够使得适合芳烃加氢饱和反应的温度范围变宽,避免因放热反应导致局部温度过高而不利于芳烃的加氢饱和。Naranov 等[12]在Ru基催化剂上探究反应条件(氢压3~7MPa、温度220~320℃、反应时间0.5~3h)对萘加氢反应的转化率和产物选择性的影响,结果表明,增大压力能够提升十氢萘的选择性。而在探究反应温度的影响时发现,240℃时产物十氢萘的收率最高,这是因为低温情况下反应速率较慢,高温下全氢产物的选择性受热力学限制,同时也会有裂化产物生成。因此,操作条件的选择要结合热力学、动力学以及反应网络等多方面因素综合考虑,优选出适宜的稠环芳烃加氢饱和反应条件[13]。
稠环芳烃加氢饱和反应的难点与其特点密不可分。Beltramone等[8,10]研究发现,在稠环芳烃的加氢饱和过程中,所含芳香环个数不同的产物之间存在竞争吸附,其中菲的反应活性与萘比较接近,两者皆远高于四氢萘,且各反应物吸附常数大小为:三芳香环化合物>双芳香环化合物>单芳香环化合物。此外,实验过程中会观测到中间产物四氢菲[8]、八氢菲[8]、四氢萘[14]等的选择性,随加氢反应的进行出现先增大后减小的趋势。这说明在逐环加氢过程中,并非所有的稠环芳烃分子都吸附在某一活性位点加氢至完全饱和后才脱附,其加氢至部分饱和后便可能从活性位点上脱附,与其他芳烃分子发生竞争吸附,致使中间产物的含量先增加后降低,且使末环的加氢受到抑制。针对上述问题,由于各加氢产物吸附常数存在差异,竞争吸附客观存在,可通过增加催化剂活性位点数量来缓解竞争吸附的影响,同时提高单个活性位点的活性,促使中间产物在各活性位点上充分转化为环烷烃,一定程度上避免竞争吸附的产生。
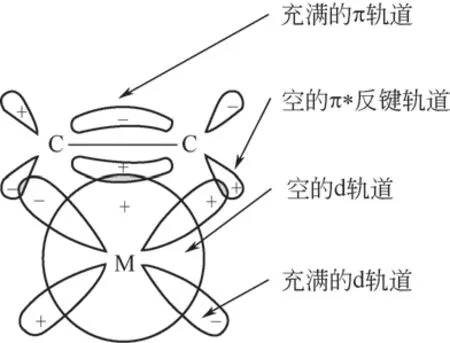
图1 π络合吸附机理[16-17]
芳烃是刚性的平面分子,倾向于平面吸附在催化剂表面[15],其在过渡金属表面的吸附机理公认为是π 络合吸附机理[16-18],如图1 所示。作用方式为芳烃的π 轨道提供π 电子给金属的d 空轨道形成σ键,再由金属充满电子的d轨道反馈部分d电子给芳烃的π*反键轨道形成d-π*反馈键,以促进成键。因此,在芳烃分子于金属表面吸附过程中,富含π电子的芳烃分子主要为电子供体、含空d轨道的活性金属为电子受体。前文分析指出,稠环芳烃分子在逐环加氢过程中共振能不断增加、空间位阻不断增大,不利于其与活性位点作用形成吸附态从而继续加氢饱和为环烷烃。但分析其吸附机理可知,吸附态的形成既与芳烃分子的属性有关,也与催化剂中活性金属的性质相关。通过调控活性金属外层电子密度形成电子部分缺失的状态,有助于富电子的稠环芳烃分子和部分加氢产物在金属表面的吸附,并抑制中间产物的脱附,促使其深度加氢饱和为环烷烃。
鉴于此,本文将对常用的稠环芳烃加氢饱和催化剂进行分类阐述,梳理各加氢催化剂活性位点的结构及其“构效关系”,结合稠环芳烃加氢饱和反应特点,对加氢催化剂的改进提出建议。
2 稠环芳烃加氢饱和催化剂
常用的稠环芳烃加氢饱和催化剂按活性组分不同,主要分为硫化物催化剂、贵金属催化剂、非贵金属催化剂以及类贵金属催化剂。其中硫化物催化剂成本较低、耐硫耐氮性较强、稳定性较好,但加氢活性相对于贵金属催化剂较弱。贵金属催化剂解离氢原子和活化芳烃分子的能力较强,低温下也具有较高的加氢饱和性能,但易被硫氮化合物毒化。而非贵金属元素中以金属Ni 加氢活性较好、成本较贵金属低,因而备受学者青睐,可单独作为加氢催化剂的活性金属。此外,一些过渡金属(Ni、Mo、Co 等)在引入C、N、P 元素后形成碳化物、氮化物、磷化物催化剂,加氢活性可媲美贵金属催化剂,被称为“类贵金属催化剂”[19]。本文将结合这四类加氢催化剂在萘、蒽、菲和芘加氢饱和反应中的应用范例,对各类催化剂活性位点的结构、性能及其调控方法逐一阐述。
2.1 硫化物催化剂
硫化物催化剂以MoS2、WS2活性相为主,其结构常用“Rim-Edge”模型来描述,其中MoS2晶体层通过范德华力堆砌形成MoS2活性相,边缘的Rim位为加氢活性位点[20]。Schachtl等[21-23]对硫化物催化剂催化菲加氢反应性能进行研究,结果表明,稠环芳烃分子倾向于以π 键的形式吸附在Mo(W)S2六棱柱结构边缘,与S-H基团发生加氢反应,如图2所示,其加氢反应速率与边缘的S-H 基团的浓度和活性有关。当MoS2催化剂中引入适量的助剂Ni时,Ni 原子会优先占据S-Edge 位,并提高其附近S-H基团的活性。
图2 稠环芳烃在Mo(W)S2表面加氢反应示意[21-23]
鉴于硫化物催化剂活性相的层状结构及边缘活性位点,除了通过提升S-H 基团的活性来促进加氢反应外,还可从活性相的分散度及其形貌结构方面入手。傅雯倩等[24]使用大分子聚合物为软模板剂水热合成出介孔的MZSM-5载体,与γ-Al2O3相比,该载体与活性相前体之间相互作用较弱,在硫化过程中可形成更多的TypeⅡ型MoS2活性相(多层结构,比TypeⅠ型加氢活性高),因此具有更高的菲加氢反应活性,其深度加氢产物(八氢菲和全氢菲之和)的选择性高达98.6%,且在含硫(二苯并噻吩)条件下稳定性优于负载的Pd 催化剂,能够连续运行60h而无明显失活。此外,作者还通过调控Mo 的负载量对活性相MoS2的构效关系进行更深入的研究,结果显示,对于TypeⅡ型MoS2活性相,提高其分散度、减少堆砌层数能暴露出更多的活性位点,提高其加氢活性。同样针对硫化物催化剂的层状结构特点和边缘加氢位点,田志坚研究员课题组[25-26]以暴露更多的边缘加氢活性位点为目标,灵活调整MoS2催化剂的制备方法,不断减少活性相MoS2堆砌层数以提升催化蒽加氢饱和反应性能,并最终通过水热合成法实现“准单层”MoS2催化剂的成功制备。
2.2 贵金属催化剂
贵金属类催化剂最突出的优势在于低温下具有优良的加氢活性及稳定性。Jacinto 等[27]将纳米Rh接枝在介孔二氧化硅载体上,其在低温低压(40℃、氢压0.4MPa)条件下也具有较高的蒽转化率(>90%),且催化剂采用磁分离循环反应6 次后没有明显的失活。但加氢产物以四氢蒽和八氢蒽为主,并未检测到全氢蒽。为了探究贵金属高活性产生的原因以及进一步提升加氢活性,先要分析稠环芳烃在金属上的吸附行为和吸附机理。Morin等[28]对稠环芳烃分子在贵金属Pt表面的吸附状态进行了研究,DFT计算结果表明,在Pt(111)表面,稠环芳烃分子和苯分子与金属Pt 形成的化学键并无本质区别。当稠环芳烃分子的中心环以桥位吸附在金属表面时,其吸附结构最为稳定,而其他环的吸附位置相对于苯环的最佳吸附位置会出现较小的横向位移,如图3所示。此外,该作者还将芳香分子和金属表面之间的电子相互作用归纳为芳烃分子的π轨道向金属表面的dxz和dyz轨道转移电子,而金属表面的dz2向芳烃分子的π*反键轨道反馈部分电子,这与前文π络合吸附机理的分析相一致。
图3 芳烃分子在Pt(111)面加氢反应示意图
因此,在加氢过程中形成金属的缺电子状态有利于稠环芳烃分子在金属表面的吸附活化,同时提高金属粒子的分散度、减小粒径能暴露出更多的金属原子,提高金属原子的利用率、增加活性位点的数量。Corma 等[29]比较了活性金属Pt 负载在不同载体(γ-Al2O3/USY/MCM-41)上催化萘加氢反应性能的变化,研究发现,分子筛较高的比表面积和规则的孔道结构使得活性金属Pt 的分散度较高、粒径较小,且分子筛表面的Brønsted酸位点促进了缺电子状态的Pt 团簇的形成,从而增强了催化剂的加氢活性和耐硫性。Williams 等[30-31]将Pt 负载在不同SiO2/Al2O3比的硅铝载体上进行萘加氢反应活性评价,结合XANES 表征发现,随着金属Pt 电子密度的降低,金属Pt与反应物之间的相互作用增强,催化剂的萘加氢活性随之提升。因此,贵金属催化剂中活性金属的分散度和缺电子状态往往是影响其加氢活性的关键因素。
2.3 非贵金属催化剂
在众多非贵金属元素中,金属Ni 与Pt、Pd 同主族,电子结构和化学性质较为接近,且价格低廉,迄今成为学者们常用的活性金属之一。与贵金属类似,Ni 具有较好的解离H2分子为氢自由基的能力[32],且DFT计算结果显示稠环芳烃分子在Ni表面吸附时存在由芳烃分子向金属Ni的电子转移[33]。
此外,Ghadami 等[33]还比较了萘分子在Ni(111)面与Pt(111)面吸附能的差异,结果显示,Ni(111)面的吸附能(4.28eV) 高于Pt(111)面的吸附能(0.51eV),因此Ni 基催化剂的加氢活性往往低于贵金属催化剂。Wu等[34]制备了一系列Ni/γ-Al2O3催化剂用于萘加氢反应体系,结果发现并没有十氢萘的生成,主要加氢产物为四氢萘。为了进一步提高Ni 基催化剂的加氢活性,需要从催化剂的结构入手,提高Ni 颗粒分散度的同时调控其电子状态。Wang 等[35]利用页硅酸镍独特的层状结构以及金属镍与载体间的强相互作用,促使镍颗粒高度分散,并在反应过程中有效抑制了活性金属的团聚。此外,页硅酸镍中不饱和的Ni 物种构成了催化剂的Lewis 酸性位点,能够吸附稠环芳烃分子,而部分页硅酸镍还原后形成金属Ni 能够解离H2,两者共同作用促使该催化剂具有优异的稠环芳烃加氢饱和性能,在循环7 次后十氢萘的选择性仍高达92%。Gong 等[36]采用一锅法水热合成出Ni-MFI-NSs 催化剂,该制备方法促使活性金属Ni通过O-Ni-O-Si-O的方式与MFI载体发生强相互作用,使得Ni于MFI纳米片外表面高度分散,同时该催化剂独特的层状结构有利于萘分子的扩散,从而实现在低Ni 负载量[4.2%(质量分数)]时具有较高的萘加氢转化率(84.9%)。但该催化剂在反应10h 后加氢活性有所下降,原因是Ni颗粒发生了团聚。
需要补充说明的是,金属Ni 的高分散度并不一定对应着高加氢活性,杨仁春等[37]认为Ni系催化剂活性组分的高度分散以及与载体间过强的相互作用可能会导致活性组分进入载体骨架,例如形成不具有加氢活性的NiAl2O4尖晶石结构,从而无法提高活性位点的数量。因此在设计提高催化剂分散度的同时,要兼顾催化剂活性位点的组成、结构和电子性质等多方面因素综合考虑。
2.4 类贵金属催化剂
为了进一步提升非贵金属的加氢活性以及耐硫耐氮性,可通过引入C、N、P 等元素形成金属碳化物、氮化物和磷化物,使其加氢活性能与贵金属相媲美,因而被称为“类贵金属催化剂”。这类新型催化剂的开发与应用激发了学者极大的兴趣。
在晶体结构方面,P 原子的半径(0.109nm)与C原子(0.071nm)和N原子(0.065nm)差别较大,因此所形成的磷化物、氮化物和碳化物在结构上有所差别。C原子和N原子能够进入过渡金属的晶格形成间隙化合物,使金属原子间距增加,晶格扩张,导致过渡金属d带收缩,费米能级态密度增加[38]。而P原子则通过占据结构单元内部空间的方式进行填充,形成三棱柱结构[39]。虽然晶体结构上有所差异,但在电子结构上,C、N、P原子的掺杂都使得电子由过渡金属向非金属转移[38,40]。将这些特殊电子结构的金属原子暴露在催化剂表面,是这类催化剂的活性能与贵金属相媲美的根本原因。
这 类 催 化 剂 中Mo2C[41]、WC[42]、Mo2N[43]、Ni2P[44]等催化加氢性能较好,特别是在含硫含氮条件下其催化性能可媲美贵金属催化剂且更稳定。但这类催化剂在反应过程中会因为活性相发生变化而导致活性下降[45],因此在制备和反应过程中维持晶相的稳定是巩固其活性的关键。以Ni2P 催化剂为例,Oyama等[46]研究发现,当初始P/Ni物质的量比为2/1时能够制备出纯相的Ni2P且在反应过程中保持不变,因而具有较好的加氢精制性能。当初始P/Ni物质的量比低于2/1时,程序升温还原后生成Ni、Ni12P5以及Ni2P的混合相,加氢活性低于纯相的Ni2P。
此外,研究表明,这类催化剂加氢活性位点的数量及性能与其形貌结构密切相关,尤其是颗粒尺寸的影响较为显著。Hu 等[47]认为减小颗粒尺寸能使得催化剂表面包含更多的非金属元素,促进电子由过渡金属向非金属转移,维持晶相的稳定并提高加氢活性。本文作者课题组[48-51]前期考察了Ce-Al2O3载体、Ni/P 物质的量比、Ni2P 的负载量、程序升温还原法和热解次磷酸盐法对Ni2P催化剂的晶相、颗粒尺寸、表面活性位数量的影响,并对催化剂的萘加氢饱和性能进行研究。结果显示,Ce的掺杂以及热解次磷酸盐法的选用都能削弱P物种与载体间的相互作用,促进Ni2P 相的生成。另外,在调控Ni2P负载量的研究过程中发现,当负载量超过最佳值时,Ni2P颗粒会发生团聚,导致颗粒尺寸大幅增加,通过CO 脉冲吸附测得的活性位点数量反而降低。除磷化物催化剂外,碳化物催化剂也有类似的性质。Jiang 等[52]的实验结果显示,当Si/Al=18时合成出的介孔NiMoC/Hβ分子筛催化剂具有最小的颗粒尺寸和较高的分散度,因而表现出最优的萘加氢活性和十氢萘选择性。此外,该研究还指出载体的酸性也是NiMoC/Hβ催化剂加氢活性的重要影响因素,其作用方式将在本文3.1节详细阐述。
总览四类加氢饱和催化剂,虽结构各异,但活性金属的电子结构、活性相的制备、分散度以及颗粒尺寸等因素往往是影响其加氢活性的关键。首先纯的活性相的合成是催化剂高活性和稳定性的前提,其次通过提高活性组分的分散度、减小其粒径可以获得更多的活性位点,缓解竞争吸附对芳烃加氢饱和反应的抑制作用,但也要综合考虑活性位点的组成、结构以及金属与载体相互作用的强弱等因素,共同提升活性位点的数量。另外,对吸附机理的分析表明,形成活性金属的缺电子状态有利于芳烃分子在金属表面的吸附活化,增强单个活性位点的加氢活性。尤其在类贵金属催化剂中,C、N、P元素的引入促使电子从活性金属向非金属发生转移,极大地提高了催化活性。因此,理想的稠环芳烃加氢饱和催化剂应具有较高的活性金属分散度以及适宜的电子状态,从而兼备较多的活性位点数量以及各活性位点较高的加氢活性。
3 促进稠环芳烃深度加氢饱和的建议
为了实现上述目标,需要结合各催化剂活性位点的结构特点和活性影响因素具体分析,通常可从活性相的制备、活性组分的电子性质以及晶粒尺寸等方面入手。其中,调节载体的酸性和添加助剂是十分有效的手段。
3.1 适当增加载体的酸性
催化剂的催化特性与载体的结构和性质紧密相关,载体较大的比表面积、适宜的孔径及孔道结构既有利于活性组分的深入负载与生长,又有利于反应物和产物的扩散[53]。另外,许多研究结果表明,载体的酸性与催化剂加氢活性关系密切,适当增加载体的酸性可以提升催化剂的加氢性能,但对其中原理的解释众多,主要包括以下三个方面。
(1)载体表面的酸性基团与活性相前体之间存在相互作用,在制备过程中影响活性组分的扩散和分布,进而影响活性组分的分散度和颗粒尺寸以及加氢活性位点的数量。Shelimov 等[54]研究发现,前体盐H2PtCl6与载体γ-Al2O3的酸性基团会通过静电作用相互连接,形成Al-O…[PtCl5(OH)]2-中间过渡态。这类相互作用广泛存在于活性组分的浸渍和离子交换过程中,致使载体的酸量和酸分布对活性组分的分散度和颗粒尺寸产生影响。Du 等[55]对混合载体的Pt-Pd双金属催化剂进行萘加氢反应性能测试,其中Al2O3-SAPO-11混合载体的酸量及其与金属的相互作用比较适宜、金属分散度较好,产物十氢萘的选择性超过了90%。Fu等[56]研究发现,与SiO2载体相比,使用双重模板法制备的微球形ZSM-5-NA 载体表面丰富的酸性羟基基团与Mo 物种之间存在强相互作用,使得钼的氧化物分散度更高,还原后活性相MoP 的粒径更小,能够提供更多的加氢活性位点,提高菲加氢活性。
(2)芳烃分子能够吸附在载体表面酸性位点上,并通过溢流氢发生加氢反应,从而提高催化剂的加氢活性[57]。Dang等[58]通过DFT方法对稠环芳烃在γ-Al2O3载体表面的吸附行为和机理进行研究,结果指出,稠环芳烃分子与载体γ-Al2O3(110)表面的羟基以及配位不饱和的铝原子之间存在色散作用,表面芳烃分子能够吸附于酸性载体表面,并倾向于平面吸附。另外,Karim 等[59]对载体表面的溢流氢现象进行了深入研究,结果表明,虽然活性氢的移动性在非还原性载体(Al2O3)表面逊色于还原性载体(TiO2)表面,但两类载体表面都存在溢流氢现象,活性氢原子在Al2O3载体表面通过“Three-coordinated aluminium centres”的方式传递。以上两方面的研究成果加深了学者对溢流氢现象以及酸性位点与金属位点之间协同作用的理解,同时相关实验研究结果进一步证实了“稠环芳烃分子吸附于载体表面并通过溢流氢加氢”的方式能够促进稠环芳烃的加氢饱和反应,并应用于催化剂的设计中。Liu 等[60]通过调控Si/Al 比制备出一系列的介孔ZSM-5 载体,并负载Pt 进行萘加氢性能评价。其中,调控Si/Al比为25、水热合成出的Pt/HZSM-5-25催化剂加氢性能较好,这与该催化剂较高的金属Pt分散度和较多的酸性位点数量有关,尤其是外表面的酸性位点数量较多。究其根本,萘分子尺寸(0.703nm×0.907nm) 较 大, 难 以 扩 散 至 孔 径(0.51nm×0.55nm 和0.52nm×0.58nm) 较小的孔道中,因此只有外表面的酸性位点才是可吸附稠环芳烃分子的加氢位点。所以尽管用NaCl 溶液处理后的Pt/HZSM-5-25 催化剂具有更多的总酸量,但外表面的酸量略低,因此加氢活性反而低于未处理的Pt/HZSM-5-25催化剂。同样利用溢流氢加氢方式,Yang等[61]将金属Pt包裹在KA沸石的微孔孔道中并与HY 和γ-Al2O3混合,只通过溢流氢与吸附在大孔道的HY 和γ-Al2O3酸性位点上的萘加氢反应,阻止了硫化物对金属Pt 的毒化,提高了催化剂的耐硫性。对于分子尺寸更大的芘,Lin 等[62]在对其加氢研究中也认为芘能够以碳正离子的形式吸附在Brønsted酸位点上,并与从附近金属Pd表面溢流的活性氢原子发生加氢反应。
(3)酸性位点与邻近金属之间的相互作用会促使金属上电荷向酸中心迁移,形成缺电子的状态。Tang 等[63]研究发现,Beta-H 载体比Al-MCM-41 载体具有更多的酸性位点和更高比例的强酸位点,小粒径的Pd 团簇与载体强酸位点紧密接触使得电子从贵金属中抽离,产生缺电子的Pd 粒子,在噻吩存在的情况下Pd/Beta-H 展现出更好的催化萘和芘加氢饱和性能。对于贵金属缺电子模型形成的机理,侯朝鹏等[64]认为:贵金属前体在离子交换过程中吸附在酸性位点上,焙烧和还原后形成的金属簇与酸性位点紧密接触使得电子脱离贵金属产生缺电子金属簇。此外,Treesukol 等[65]通过DFT 计算指出,分子筛骨架上的氧原子会提供电子给Pt原子,而B酸位点上的氢质子会剥离Pt原子中的电子,两者共同作用导致Pt原子的电子重新分配,增加s轨道电子密度的同时削弱d轨道的电子密度。
通过以上三个方面的作用,增强载体的酸性通常能提高催化剂的加氢饱和性能。此外,邓澄浩等[66]认为金属与酸性位点的协同作用能抑制分子筛酸性位点上积炭的形成,改善催化剂的稳定性。而姬宝艳等[67]则认为载体的强酸中心(如Brønsted酸)与金属间的相互作用会导致稠环芳烃的开环裂化。同理,Cui 等[68]在研究Ni2P 催化萘加氢性能时发现,当载体的酸性较强,尤其是Brønsted酸较强时,加氢活性位点Ni2P 会与酸性位点发生协同作用,导致加氢产物异构及开环裂化,并给出了可能的反应路径。可见,只有适当增加载体酸性才能促进芳烃的加氢饱和,过量的酸性则会导致加氢产物的开环裂化。
3.2 添加助剂以提高分散度、减小粒径
通过添加助剂可以改变活性组分与载体间相互作用的强弱,提高活性组分的分散度、减小其颗粒尺寸,从而获得更多的加氢活性位点。但实际过程中需要针对不同催化剂加氢位点的结构来进行调控。Zhang 等[69]在催化剂的制备过程中加入柠檬酸(CA),形成的CA-Ni 复合物与载体的相互作用较强,还原得到的Ni2P 活性相分散度较好、粒径较小,加氢活性较高,反应56h后菲的转化率没有明显下降。Qiu等[70]在Ni(NO3)2中加入适量的乙二醇共浸渍到MCM-41 载体上,同样增强了Ni 物种与载体间的相互作用,得到分散度更高、粒径更小的NiO颗粒,在萘加氢活性测试中,十氢萘的选择性显著提升。Vargas-Villagrán 等[71]在制备Ni/SBA-15催化剂时添加助剂EDTA,形成[Ni(EDTA)]2-复合物,制备出的Ni 颗粒粒径(2.7~6.2nm)更小、分散度更高,同时也提升了载体酸性。两者共同作用同样提高了十氢萘的选择性。除非金属助剂外,金属助剂也可促进活性组分的形成与分散,获得更多的活性位点。在MoS2催化剂中,Schachtl 等[21]研究发现,助剂Ni的存在能够削弱Mo物种与载体的相互作用,促进氧化物前体中八面体Mo物种的形成,降低Mo物种的还原温度并提高活性相MoS2的分散度,增加加氢位点的数量。但研究也表明,过量的Ni则会形成无加氢活性的NiSx团簇,不利于催化剂活性的提升。
3.3 添加助剂以形成缺电子状态
以促进芳烃的吸附活化为目标,通过添加助剂可以实现活性金属电子结构的调控,形成电荷局部缺失的金属位。本文2.4 节分析表明,非金属元素如C、N、P 的掺入能够促使活性金属上的电子转移出去,形成缺电子状态从而提升其加氢活性,在此不再赘述。除此之外,利用双金属之间存在的相互作用,通过引入第二金属组分也能调控活性金属外层电子密度,发挥类似的作用。Navarro 等[72]研究发现,在含硫的情况下PtPd 双金属催化萘和甲苯加氢饱和反应性能远高于单金属的Pt 和Pd。从表征结果来看,PtPd 双金属间的相互作用促使Pt形成缺电子的状态,因而具备更高的耐硫性和加氢活性。此外,Matsubayashi 等[73]通过EXAFS 表征也发现PtPd 双金属间会形成Pd-Pt 键。类似的,以AlCl3为助剂引入金属Al 能够降低活性金属Pt 的电子密度,形成缺电子的Ptδ+从而提高催化剂的加氢活性和耐硫性[74]。对于双金属间的相互作用及其活性变化规律,Nørskov 等[75-76]通过d带中心理论来阐释和总结,相关计算结果也可为双金属催化剂设计和选择提供指导。
4 结语
随着人们环保意识的增强和对燃油品质的重视,将油品中稠环芳烃转化为环烷烃仍是备受关注的研究热点之一。受共振能、空间位阻和竞争吸附影响,稠环芳烃在加氢饱和过程中末环的吸附活化困难,导致其深度加氢饱和转化率往往不高。而研制高效的稠环芳烃加氢饱和催化剂是解决该难点的有效途径。
基于稠环芳烃加氢饱和反应特点和吸附机理的分析,除了适宜的加氢反应条件外,提升催化剂加氢活性的关键在于提升活性位点的数量和调控活性金属的电子状态。针对四类不同的稠环芳烃加氢饱和催化剂,本文分别阐述了它们活性相的结构和影响其活性的主要因素,并根据不同催化剂的结构特点指出了其加氢活性提升方法。分析表明,在确保孔道结构利于扩散、比表面积适宜的情况下,调控载体的酸性是提升催化剂加氢活性的有力举措。除此之外,采用添加助剂的方法同样能够在活性相的结构、分散度以及金属的电子结构等方面产生促进作用,增加活性位点的数量或者形成活性金属的缺电子状态,进一步提升加氢活性。
