非均相催化剂催化5-羟甲基糠醛氢解制备2,5-二甲基呋喃研究进展
2021-03-08王彤安华良李芳薛伟王延吉
王彤,安华良,2,李芳,2,薛伟,2,王延吉,2
(1 河北工业大学化工学院,河北省绿色化工与高效节能重点实验室,天津300130;2 天津市本质安全化工技术重点实验室,天津300130)
随着化石能源的日益短缺及人们对全球气候变化(温室效应)与日俱增的担忧和关注,迫切需要开发低成本、安全、环保、可持续的新能源来替代传统化石能源[1]。生物质是一种产量巨大的可持续有机碳资源,全球年产约1.7×1011t[2]。生物质主要来源于植物的光合作用,其中,木质纤维素是储量最丰富的非粮生物质,具有很大利用潜力。在木质纤维素的利用途径中,将其转化为高质量的液体生物燃料和高附加值的化学品无疑是最具吸引力的方法之一[3-5]。木质纤维素的高效转化利用离不开催化剂的催化作用,设计高效、绿色的催化体系实现可再生生物质资源的合理利用是当前热点研究领域之一[6-8]。
2,5-二甲基呋喃(DMF)是一种非常有应用前景的可再生液体燃料,具有优良的物理化学性质。相较于乙醇、丁醇,DMF 有更高的能量密度(31.5MJ/L),与汽油(35MJ/L)和柴油(33MJ/L)类似;同时,DMF 具有较高的辛烷值(RON=119),作为燃料的效率更高。此外,DMF 的沸点介于乙醇和丁醇之间,具有合适的气化性能,有利于降低发动机进气道的空气阻力,能够满足发动机低温启动的需求;并且,DMF 比乙醇更容易与汽油混合,更有利于运输和储存。这些特性使得DMF成为生物乙醇等液体燃料的理想替代品[9]。
5-羟甲基糠醛(HMF)可由纤维素水解制得,是连接生物质和精细化工化学品及石油基资源的重要桥梁,被称为“沉睡的巨人”[10]。由于HMF 分子中含有丰富的官能团(呋喃环、醛基和羟甲基),其化学性质十分活泼,可以衍生出许多高附加值的化学品(图1)。如氧化生成2,5-呋喃二甲醛(DFF)、5-羟甲基-2-糠酸(HMFA)、2,5-呋喃二甲酸(FDCA)、己二酸(HDA);氢化/氢解生成2,5-二羟甲基呋喃(DHMF)、2,5-二甲基呋喃(DMF)、2,5-二甲基四氢呋喃(DMTHF)、1,6-己二醇(HDO);还可以转化成乙酰丙酸(LA)、γ-戊内酯(GVL)等[11-16]。因此,要实现由HMF高选择性制备DMF,精确设计、制备有效的靶向催化剂是十分必要的。

图1 由HMF合成的各种衍生物
基于此,本文从催化剂的角度,依据贵金属和非贵金属催化剂进行分类,系统综述了近年来非均相催化HMF 氢解制备DMF 的最新研究进展,如表1 所示。针对目前研究中存在的局限性和问题,指出了催化剂及反应体系的研究方向,以期为催化剂的进一步设计开发提供参考,为降低DMF 生产成本及实现安全生产过程提供理论支撑。此外,本文还对DMF 工业化生产前景进行了展望。
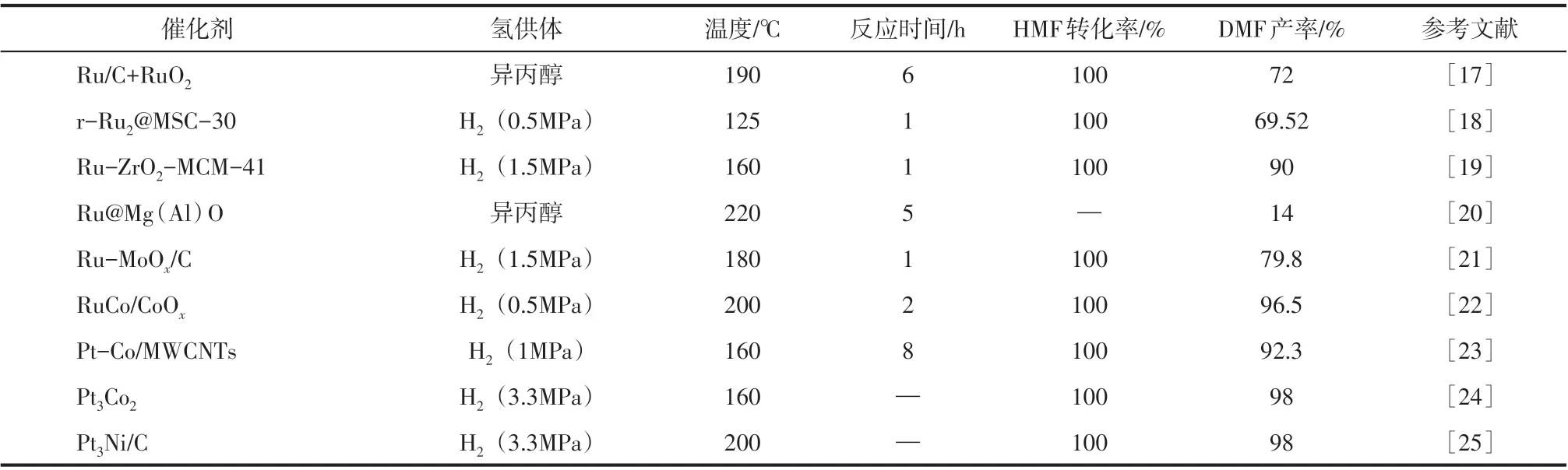
表1 用于HMF氢解制备DMF的非均相催化剂
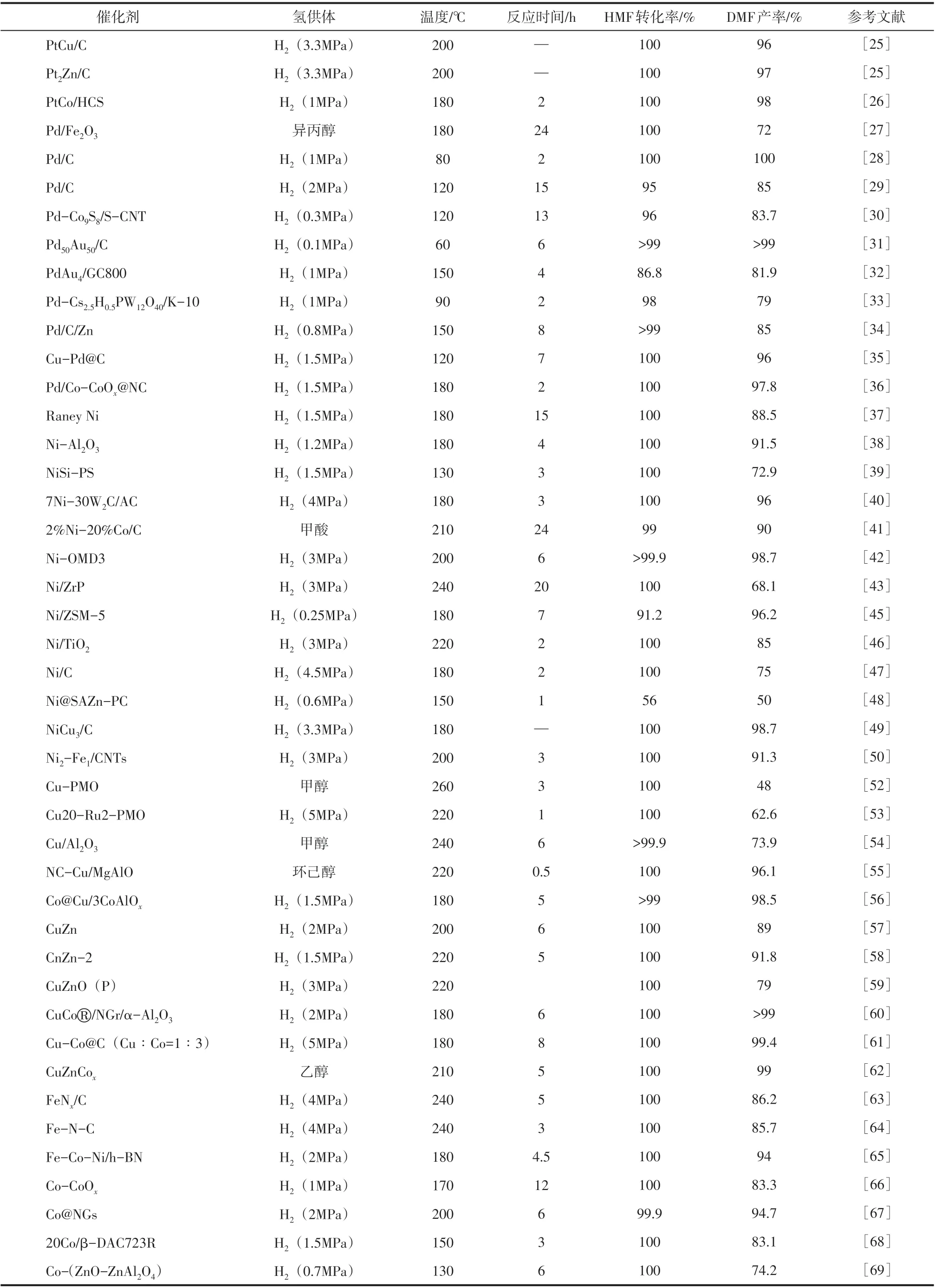
续表1
1 贵金属催化剂上HMF氢解制备DMF
HMF 氢解制备DMF 常用的贵金属催化剂有Ru、Pt、Pd等,贵金属因d电子轨道未填满而极易吸附反应底物,有利于形成中间“活性反应物”,通常在相对温和的反应条件下具有较高的催化活性和选择性。
1.1 Ru基催化剂
Ru 元素的d 带较宽,对C==O 加氢反应的选择性一般较高,且价格相对便宜。Jae 等[17]采用商业Ru/C 和RuO2混合物作为催化剂,探究其组成与HMF 氢解生成DMF 催化性能之间的构效关系。单独以RuO2为催化剂时,主要生成2,5-二羟甲基呋喃(DHMF),及其与异丙醇醚化反应得到的一些中间产物;而单独以Ru/C 为催化剂时,可以生成DMF,但选择性(30%)相对较低;当二者同时作为催化剂时,DMF 选择性大幅度提高至70%,表明RuO2和Ru/C间的协同作用至关重要。Tzeng等[18]分别以商业多孔碳MSC-30、MSP-20、介孔碳CMK-3 及无孔碳BP-280 作载体,负载Ru 催化HMF氢解反应。0.5MPa H2、125℃反应1h,催化活性 顺 序 为 r-Ru2@MSC-30>r-Ru2@MSP-20>r-Ru2@CMK-3>r-Ru2@BP-280。DMF 收 率 最 高 达69.52%,HMF 完全转化;而r-Ru2@BP-280 催化剂,HMF转化率仅有35.48%,且主要生成DHMF。催化性能的差异可能是由于碳材料本征性质不同而导致热还原过程中Ru 与碳表面相互作用不同,进而对Ru NPs 的形成及其电子结构产生影响。Raut等[19]制备了2%(质量分数)Ru-ZrO2-MCM-41 催化剂,在160℃和1.5MPa H2条件下反应1h,DMF收率达90%,催化剂循环使用4 次后收率略有下降。Nagpure 等[20]采用含Ru 水滑石衍生的Ru@Mg(Al)O为催化剂,以异丙醇作氢源,在220℃反应5h,DMF 收率较低,仅为14%。Yang 等[21]采用浸渍法,制备了兼具金属活性位和酸性位点的双功能Ru-MoOx/C 催化剂,其中金属位点负责C==O加氢及C—O 键断裂后的加氢,而酸性位点主要负责C—O 键的断裂氢解。Ru 与MoOx的协同催化,使得反应1h 即可高选择性生成DMF,收率为79.8%。该催化剂循环使用3 次后逐渐失活,但经H2还原再生后活性基本恢复。有趣的是,作为对比,发现单独以Ru/C 作催化剂时,DMF 则易过度加氢生成DMTHF,选择性较低;而仅使用MoOx/C作催化剂时,与Ru-MoOx的催化反应路径不同。
Gao 等[22]通过简单有效的封装法,制备了蒲公英状CoOx负载RuCo双金属催化剂,并提出了HMF在RuCo/CoOx催化剂上氢解的反应机理,如图2 所示。双金属RuCo NPs 独特的蒲公英状结构与载体表面丰富的表面缺陷(氧空位、Co2+物种)相互作用,促进了氢气的解离,增强了氢溢流效应,有利于HMF 分子中C==O 键的吸附和活化,使DMF 收率较高。在高HMF/Ru物质的量比(252.7)下仅仅反应2h,DMF产率就可达到96.5%;并且,催化剂特殊的封装结构及RuCo NPs 和CoOx载体间强的相互作用能够明显抑制Ru的流失,循环使用5次后,DMF收率仅下降1%。

图2 RuCo/CoOx制备示意图及RuCo/CoOx催化HMF氢解生成DMF反应机理[22]
由此可见,不同价态Ru 活性组分间的协同及金属与载体之间的相互作用是影响负载Ru 催化剂性能的重要因素。选择合适的催化剂助剂和载体,调控催化位点的微观几何结构和电子结构,有助于提高HMF氢解生成DMF的收率。
1.2 Pt基催化剂
Pt 与Ru 相似,对C==O 加氢的选择性较高。Wang等[23]通过原子层沉积(ALD)法制备了Pt-Co/MWCNTs 双金属催化剂,在160℃和1MPa H2条件下反应8h,DMF收率可达92.3%。相较于单金属Pt催化剂,反应所需H2压力大大降低。实验表征结合密度泛函理论(DFT)计算表明,增强Pt-Co 双金属NPs的电荷密度可以促进催化剂吸附C==O键,从而提高DMF 的选择性,其中多壁碳纳米管(MWCNT)在Pt 与Co NPs 之间的电荷转移过程起到重要的桥梁作用。但是,由于Co 与载体之间的相互作用力较弱,导致Co 在反应过程中的浸出,催化剂循环稳定性不佳。Luo 等[24]制备了一种具有独特核壳结构的催化剂Pt3Co2,其核由Pt-Co 合金组成,壳层是具有蜂窝状结构的Co3O2单层。DFT计算表明,壳层Co3O2单层对呋喃环吸附较弱,可以抑制过度氢化和呋喃环的开环形成副产物,提高对DMF 的选择性。随后,其课题组[25]使用溶剂热法制备了Pt3Ni/C、PtCu/C 和Pt2Zn/C 一系列双金属合金催化剂,并在连续流反应器中对这些催化剂进行了活性评价。在3.3MPa H2压力下,所有催化剂生成DMF 的收率均高于95%,表明第二金属的引入可提高Pt催化HMF氢解制备DMF的性能。Wang等[26]制备了空心碳球包覆的双金属PtCo/HCS 催化剂,以正丁醇为反应溶剂,在180℃和1MPa H2压力下反应2h,DMF收率可达98%。
1.3 Pd基催化剂
Pd元素的d带宽度相对较窄,与π键之间存在较强的相互作用,通常更有利于呋喃环的氢化。Scholz 等[27]制备了Pd/Fe2O3催化剂,以异丙醇作为氢供体,在180℃反应24h,HMF 可以完全转化,DMF 收率为72%。Chatterjee 等[28]选用商业Pd/C 作催化剂,探究HMF在超临界二氧化碳-水反应介质中的氢解性能。实验数据表明,氢气压力、反应温度以及二氧化碳与水的物质的量比对DMF 选择性有重要影响,同时由于CO2、H2O 在反应体系中的共存使得反应介质呈酸性,更有利于DMF的生成。在80℃反应2h,DMF收率达100%。Mitra 等[29]使用商业Pd/C催化剂,通过在反应体系中加入HCOOH或CH3COOH,在120℃和0.2MPa H2条件下反应15h,DMF 收率可达85%。弱酸的加入可以实现在较低的H2压力下得到较高的DMF 收率,其中HCOOH还可作为H2的温和来源,同时抑制脱羰和呋喃环氢化反应的进行。
与单金属催化剂相比,双金属协同催化通常具有更佳的反应性能。Liao 等[30]以硫改性碳纳米管(S-CNT)为载体,采用浸渍法制备了Pd-Co 双金属催化剂。硫改性载体为金属纳米粒子在碳纳米管表面成核提供了高度分散的金属锚定位点,Pd与Co9S8纳米颗粒之间的协同作用使得Pd-Co9S8/S-CNT 具有显著的氢解/加氢性能。其中Pd 主要起催化醛基加氢的作用,而Co9S8有利于羟基的氢解。在120℃和0.3MPa H2条件下反应13h,HMF转化率为96%,DMF选择性为83.7%。Nishimura等[31]采用NaBH4还原法,制备了具有不同Pd/Au 物质的量比的PdxAuy/C 催化剂。当向反应体系中加入HCl,H2压力为常压,在60℃反应6h,使用Pd50Au50/C 作为催化剂,HMF可实现完全转化,DMF收率达96%。采用相同方法制备了石墨化碳(GC)负载PdAu催化剂[32],当反应体系中不添加HCl时,在相对较高的温度和压力(150℃、1MPa H2)下,DMF收率为81.9%。综上所述,酸对HMF氢解反应具有重要的促进作用,可以提高DMF 的选择性,同时使反应条件趋于温和;但是,向反应体系中添加酸通常会带来反应器腐蚀及废弃物产生等问题,不利于该方法的产业化。基于此,Gawade 等[33]以酸性黏土K-10 作载体,采用两步浸渍制备了2%Pd-20%Cs2.5H0.5PW12O40/K-10 催化剂,在90℃和1MPa H2条件下反应2h,DMF 收率达81%,可能的反应机理如图3 所示。其中,Cs2.5H0.5PW12O40/K-10 提供的酸性位点对DHMF 经由5-甲基糠醇(MFA)快速氢解生成DMF 起重要作用。Saha 等[34]采用路易斯酸ZnⅡ和Pd/C 组成的双金属催化剂对HMF 进行氢解反应生成DMF,得到了高HMF 转化率(99%)和高DMF 选择性(85%);而在相同的反应条件下,Pd/C 催化剂上DMF 收率不到60%。这表明反应体系中路易斯酸的存在对HMF 加氢脱氧具有促进作用。Sarkar 等[35]通过热解浸渍Pd 的Cu-MOF(HKUST-1),制备了碳包覆的Cu-Pd 双金属纳米粒子Cu-Pd@C 催化剂,经H2还原后展示出优异的催化性能。在120℃和1.5MPa H2条件下反应7h,DMF 收率为96%。以HKUST-1 为牺牲模板衍生的碳骨架可以稳定均匀分散的金属纳米粒子,提高催化剂的稳定性。Shang 等[36]制备了Pd/Co-CoOx@NC催化剂,在HMF氢解生成DMF反应中,DMF选择性高达97.8%。含Co-CoOx的N 掺杂多孔碳载体对Pd 起到锚定和高分散作用,同时载体表面丰富的表面缺陷增强了催化剂对底物的吸附及表面电子转移能力,促进H2溢流和HMF氢解级联反应的进行,反应机理如图4所示。
2 非贵金属催化剂上HMF氢解制备DMF
相较于贵金属催化剂,非贵金属催化剂因成本较低而备受关注。Ni、Cu是最常见的加氢催化剂,最近的研究表明,Co、Fe 基催化剂对HMF 氢解制备DMF反应也具有不错的催化效果。

图3 Pd-CsDTP/K-10催化HMF氢解生成DMF的机理[33]
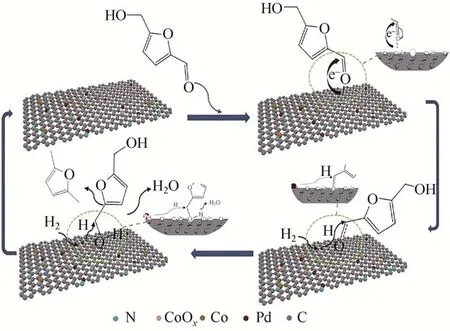
图4 Pd/Co-CoOx@NC催化HMF氢解生成DMF可能的反应机理[36]
2.1 Ni基催化剂
Ni 是一种过渡金属,其加氢活性比Fe、Co高,但不如Pt、Pd 等贵金属。正因为如此,具有中等活性的Ni 可以避免产物的进一步加氢,并可通过改变反应条件调控反应产物的选择性,是加氢反应常用的催化剂。Kong等[37-39]考察了一系列Ni基催化剂催化HMF氢解反应性能,包括Raney Ni、Ni-Al2O3和NiSi-PS(Ni NPs 嵌入层状硅酸镍)。以Raney Ni 为催化剂时,在100℃反应主要生成2,5-二羟甲基四氢呋喃(DHTHF),选择性高达96%,表明较低温度下只发生氢化反应,不具备加氢脱氧能力;而当反应温度较高(180℃)时,产物主要为DMF。由此可以看出,高温更有利于CH2—OH的氢解。对于Ni-Al2O3催化剂,通过改变焙烧温度,精细调控催化剂表面金属位点及酸性位点,并优化反应工艺参数,即可实现DMF、2,5-二甲基四氢呋喃(DMTHF)和DHTHF 的可控合成,且收率均高于90%。NiSi-PS 催化剂在低温(130~150℃) 条 件 下 可 将HMF 高 效 氢 解 成DMF 和DMTHF,与浸渍法制备的Ni/SiO2相比,NiSi-PS催化的HMF 转化率是Ni/SiO2的2 倍,氢解速率是Ni/SiO2的3倍。NiSi-PS催化剂优异的催化性能源于高度分散的Ni 纳米颗粒与位于层状硅酸盐结构上的配位不饱和Ni(Ⅱ)位点所形成的大量酸性位点的协同作用。Huang 等[40]以碳材料作为载体,制备了一系列不同金属配比的Ni-W2C/AC 催化剂,其中7Ni-30W2C/AC 催化剂在180℃和4MPa H2条件下,DMF 收率为96%。催化剂中Ni 活性组分表现出良好的加氢能力,而W2C 则表现出良好的脱氧能力,两者协同作用使DMF 具有较高的选择性。Yang等[41]制备了2%Ni-20%Co/C 催化剂,以甲酸为氢供体,在210℃反应24h,DMF 收率为90%;而使用1.5MPa H2作氢源时,DMF 选择性低,收率仅为61%。Goyal 等[42]制备了以介孔富氮碳材料为载体担载的Ni基催化剂Ni-OMD,实现了Ni NPs的高分散[3%(质量分数)Ni NPs<5nm]。以水为反应溶剂即可实现HMF 氢解生成DMF,且随着反应温度和H2压力的升高,Ni-OMD 对DMF 的选择性增加;同时,由于衬底的N 原子和Ni 原子之间存在非常强的相互作用,在循环实验中表现出良好的稳定性。Zhu 等[43]采用离子交换法制备了层状α-ZrP 担载Ni 基催化剂Ni/ZrP,在5MPa H2和240℃条件下反应20h,DMF 收率为68.1%。然而,从工业化生产及经济效率考虑,高温高压的反应条件及相对较低的收率使其应用具有一定局限性。Sun 等[44]以36Ni-12Cu/SBA-15 为催化剂,HCOOH 为氢源,HMF 与HCOOH 酯化的中间产物5-甲酰基糠醛(FMF)为底物(FMF与HMF之间存在可逆平衡),在220℃反应5h,DMF收率达71.0%。Guo等[45]采用简单的固相研磨法制备了Ni/ZSM-5 催化剂,催化剂表面适当比例的金属位点和酸性位点的协同作用对于选择性氢解生成DMF 非常重要。在最佳实验条件(180℃、0.25MPa、7h)下,可获得91.2%的HMF 转化率和96.2%的DMF 选择性。Przydacz 等[46]制备了不同晶型TiO2担载的镍纳米粒子催化剂,发现Ni 晶粒尺寸及载体酸性影响HMF 反应路径及产物分布。金红石型TiO2催化剂中Ni-Ti 之间相互作用较强,易过度加氢生成DMTHF;而大比表面积的锐钛矿型TiO2上Ni NPs 较小,主要生成DMF。此 外, Ni/C[47]、 Ni@SAZn-PC[48]、 NiCu3/C[49]和Ni2-Fe1/CNTs[50]等催化剂也被用于釜式及连续流反应器中的HMF 氢解反应,对DMF 均具有很高的选择性。
2.2 Cu基催化剂
除Ni 基催化剂外,Cu 基催化剂因其独特的催化反应性能和较低的成本一直以来备受关注。在HMF 氢解成DMF 的反应中,活性金属Cu 由于其d轨道电子排布能对C原子产生强排斥,可以优先活化C==O键,保护C==C键不被加氢,并最大程度地减少脱羰作用。Román-Leshkov 等[51]首次提出以生物质衍生物平台分子制备液体燃料,采用CuCrO4作催化剂,在200℃下反应10h,HMF可完全转化,DMF 收率为61%。Hansen 等[52]报道了以Cu-PMO(多孔金属氧化物)为催化剂,在超临界甲醇中进行HMF的选择性氢解。260℃下反应3h,HMF完全转化,DMF 收率为48%;Kumalaputri 等[53]制备了Ru掺杂的Cu20-Ru2-PMO催化剂,可使HMF完全转化,DMF 收率为62.6%。Zhang 等[54]使用甲醇作为氢供体,考察不同载体制备的催化剂Cu/Al2O3、Cu/ZnO、Cu/ZrO2及Cu/CeO2对HMF 氢解反应活性的影响。其中,Cu/Al2O3催化剂中Cu 晶粒尺寸最小,载体酸性最强,因此甲醇原位制氢活性最高,催化HMF氢解性能最优。在240℃下反应6h,DMF收率为73.9%。Gao 等[55]通过简单热分解CuMgAl-LDH/三聚氰胺杂化物前体的方法,制备了价格低廉的氮掺杂碳(NC)修饰的Cu基催化剂(NC-Cu/MgAlO)。以环己醇作为氢供体和溶剂,用于HMF转移氢解反应,如图5 所示。NC 的引入使得催化剂表面Lewis碱性位点增多,同时调变Cu物种的电子状态,有助于Cu+的形成,而Cu0-Cu+的平衡及其与Lewis 碱性位间的协同效应增强了催化剂对环己醇脱氢和HMF 氢解性能。在220℃反应30min,HMF 即完全转化,DMF 选择性高达96.1%。并且,N 的引入增强了Cu 与载体之间的相互作用,催化剂重复使用5 次,依然表现出良好的催化性能。Wang 等[56]通过还原CuMgAl-LDHs 和CuCoAl-LDHs水滑石前体,构筑了Cu-MgAlOx(惰性载体)和Cu-CoAlOx(活性载体)两种具有不同活性界面的催化剂,发现活性金属与氧化物载体间不同的表界面结构对反应路径及产物分布有显著影响。Cu/MgAlOx仅对C==O键加氢有活性,主要生成DHMF,收率为92.7%;Co@Cu/3CoAlOx可进一步催化氢解生成DMF,收率高达98.5%;而通过调节Cu/Co 比例(改变Cu-Co界面)制备的Co@Cu/5CoAlOx催化剂,则可生成DMF 深度加氢产物2,5-二甲基四氢呋喃(DMTHF),收率为83.6%。
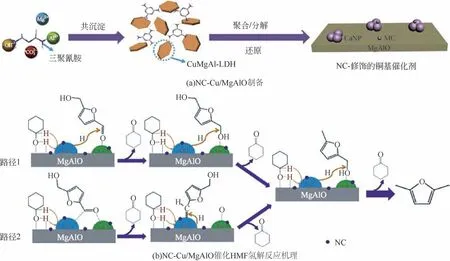
图5 NC-Cu/MgAlO制备及NC-Cu/MgAlO催化HMF氢解反应机理[55]
Zn 是铜基催化剂中常用的掺杂剂之一,对于提高Cu 分散度有重要作用。Bottari 等[57]制备了CuZn纳米合金催化剂,在220℃反应可获得较高的DMF 收率。Zhu 等[58]采用共沉淀法制备了Cu-ZnO双金属催化剂,当铜锌物质的量比为2(CuZn-2)时,由于催化剂表面Cu 含量最高,酸度适中,且具有良好的微观结构,其反应活性最高。通过简单调变反应温度和反应时间,即可实现调控DMF 和DHMF 的靶向合成。然而催化剂在第二次使用时,HMF转化率下降了40%,DMF收率小于20%,表现出较差的稳定性。Brzezińska等[59]采用光辅助合成法制备了CuZnO(P)催化剂,在220℃和3MPa H2压条件下反应5h,HMF完全转化,DMF收率为79%;而采用浸渍法制备的CuZnO(Ⅰ)在相同反应条件下,主要生成DHMF,收率为93%。导致产物差异的原因在于催化剂制备过程中,ZnO在辐照下易发生表面光腐蚀,可在CuZnO(P)上形成Lewis 酸位点(Cu-ZnO 界面位点的形成),有助于DHMF 进一步氢解。然而由于CuZnO(P)上形成的Cu 纳米颗粒尺寸较小,更易使催化剂因积炭导致失活。
此外,Co 是金属催化剂体系合适的促进剂,Cu-Co双金属之间的协同效应可以提升催化剂的催化性能。Guo 等[60]以N 掺杂石墨烯修饰的氧化铝为载体,担载CuCo 纳米粒子,制备了CuCo®/NGr/α-Al2O3催化剂。在180℃和2MPa H2压条件下反应16h,DMF 收率达99%。Chen 等[61]通过在氩气中热解聚乙二醇沉积的铜钴氧化物,制备了碳包覆双金属CuCo纳米催化剂。聚乙二醇既是壳层碳的来源,也是金属物种的还原剂;CuCo 双金属协同催化使其在HMF 氢解过程中具有极佳的选择性和活性,在5MPa H2压和180℃条件下反应8h,DMF 收率可达99.4%;同时碳壳层的包覆可保护双金属纳米颗粒免于氧化和失活,6次循环使用后仍具有良好的稳定性。Zhang 等[62]采用共沉淀法制备了三元CuZnCox催化剂,以乙醇作氢源,在210℃下反应5h,DMF收率高达99%。
2.3 Fe基催化剂
Fe 基催化剂在加氢/氢解反应中的应用相对较少。Li 等[63]分别以活性炭和1,10-邻菲咯啉为碳、氮源,采用高温热解法制备了铁基催化剂FeNx/C,用于HMF 氢解合成DMF 反应。在正丁醇中240℃下反应5h,HMF 转化率为100%,DMF 选择性为86.2%。随后,通过在活性炭上热解三聚氰胺和乙酰丙酮铁,制备了Fe-N-C 催化剂,同样取得了不错的催化效果[64]。Chen等[65]以成本低且在极端物理化学条件下稳定性良好的六方氮化硼(h-BN)为载体,采用沉积沉淀法制备了三组分非贵金属负载催化剂Fe-Co-Ni/h-BN,通过一系列表征可以确定铁镍形成合金,同时铁镍合金和钴物种在氮化硼表面均匀分散。在最优反应条件下,DMF 收率高达94%。催化剂循环使用10 次后,HMF 仍可完全转化,DMF收率仅下降12%。
2.4 Co基催化剂
Co 除了用作金属催化剂的促进剂外,其自身单独也可作为金属活性中心,具备催化活性。Li等[66]通过沉淀法制备了Co3O4,通过控制还原温度得到兼具金属位点(Co) 和路易斯酸性位点(CoOx)的双功能催化剂Co-CoOx。当还原温度为400℃时,Co-CoOx催化剂中负责解离活性氢及醛基加氢的金属活性位点与活化C—O 键氢解断裂的酸性位点的量达到一定的平衡,协同催化使得其在170℃反应12h,HMF 即可完全转化,DMF 收率为83.3%。催化剂在6 次循环使用后仍可保持75%的DMF 收率,表明其作为一种低成本催化剂在生物质转化方面具有潜在的应用价值。最近,Wang等[67]利用莫特-肖特基界面效应调控含氮碳材料和金属活性纳米粒子的界面电子重排,制备了一种新型、具有封装结构的高效碳氮复合催化材料Co@NGs。石墨烯壳层可以有效地抑制Co NPs的团聚和浸出,并保持活性金属Co 的良好催化活性和稳定性。通过提高碳衬底上氮掺杂含量可使Co@NGs 功函数增加,从而提高Co@NGs 异质结界面的整流响应,提升催化界面性能。实验结果表明,金属Co 电子转移至掺杂N 致使其自身电子密度下降,而电子缺陷的Co有利于HMF选择性催化加氢制备DMF。最佳Co@NGs催化剂表现出较高的HMF 转 化 率(接 近100%) 和DMF 选 择 性(94.7%),远优于传统的氮掺杂碳负载Co 催化剂。Chen 等[68]制备了Hβ 分子筛担载的Co 基催化剂Co/β-DA,通过调节焙烧温度调变催化剂表面Co物种的微环境和金属颗粒大小。450℃及以上温度焙烧时,Co 物种以孤立位点形式存在于沸石骨架,同时以CoOx形式存在于沸石表面或稳定在沸石孔道内,经氢气还原后,大部分Co 物种被还原成相应的金属Co。以20Co/β-DAC723R为催化剂,150℃、1.5MPa H2压下反应3h,HMF 可完全转化,DMF收率为83.1%。An 等[69]采用尿素沉淀法制备了Co-(ZnO-ZnAl2O4) NPs,在较温和的条件(130℃,0.7MPa H2压)下反应24h,DMF收率为74.2%。
总体而言,由于非贵金属催化剂的活性比贵金属催化剂(包括Ru、Pt、Pd)低,通常需要较高的反应温度和压力;而贵金属催化剂则由于活性较高,如何避免HMF 氢解成DMF 过程中的进一步加氢或其他副反应是值得重视的问题。另外,对于不同的反应器,连续流反应器比间歇式反应器需要更高的H2压力。
3 反应机理
HMF 分子中呋喃环上含有醛基和羟甲基,根据活化氢攻击HMF 上C==O 键和C—O 键的先后顺序,其氢解过程可以分为两种反应路径:路径1是HMF 上的醛基先加氢生成2,5-二羟甲基呋喃(DHMF),然后呋喃环上的羟基进行两步氢解,经由5-甲基糠醇(5-MFA)反应中间体生成DMF;路径2 是HMF 上的羟甲基先发生氢解生成5-甲基糠醛(5-MF),随后5-MF 上的醛基加氢生成5-MFA,最后5-MFA 上的羟基进一步氢解生成DMF,反应网络如图6 所示。尽管反应路径1 所需活化能远低于反应路径2,但在有些催化体系中反应按路径2 进行,这可能跟HMF 在催化剂表面的吸附及催化活性位点有关。因此现有机理解释尚不具备普遍适用性[70],通过精确调控催化活性位点调变反应路径还有待进一步深入研究。
4 总结与展望
综上所述,贵金属催化剂在HMF 氢解反应中具有较强的解离氢气和转移氢的能力,对生成DMF 具有很高的催化活性,但是由于价格昂贵,限制了其大规模应用。而非贵金属催化剂尽管价格上占有一定优势,但催化活性相对较低,大部分循环稳定性较差。为实现DMF 的工业化生产,对今后的研究提出以下几点建议。
(1)设计合成高效的非贵金属催化剂和低含量的贵金属催化剂迫在眉睫。近年来,单原子催化剂(SACs)以其优异的催化活性引起了人们的广泛关注,能否设计贵金属基SACs,尤其是非贵金属SACs,将其应用于HMF 氢解反应,解决催化剂成本高及循环稳定性较差的问题,是很有潜力且重要的研究方向。另外,目前大多数研究选择分子H2作为氢供体,催化转移氢化的研究还是相对较少,实现以环境友好氢供体为氢源,达到与H2同样的反应效率,从本质安全评估角度是非常有前景的。
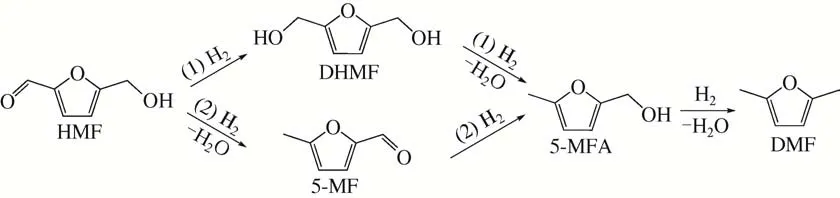
图6 HMF转化为DMF的反应路径
(2)采用更多先进的原位表征技术并结合密度泛函理论(DFT)计算,深入了解非均相催化剂界面的催化位点特性,从而评估每种活性位点的本征催化活性,揭示不同催化位点上的反应机理,以期真正实现为优化设计催化剂提供理论指导。
(3)设计制备方法简单且价格合理的双功能催化剂,实现由生物质(糖类、纤维素和木质素)一锅法催化转化制备DMF,可以大大提高DMF 的生产效率。此外,研究建立有效的分离技术,将DMF 从复杂的副产品中分离出来是其进一步应用和实现工业化生产的前提。
