UFLC-Q-TOF-MS/MS结合指纹图谱定性评价菝葜(金刚藤)药材质量的研究
2021-03-05龙丽冯海兴王灿茂杨琴吴馥凌侯连兵
龙丽,冯海兴,王灿茂,杨琴,吴馥凌,侯连兵
(南方医科大学南方医院,广东 广州 510515)
菝葜(SmilaxchinaL.)又名金刚藤,是百合科植物菝葜的干燥根茎,主要分布于长江以南的多个省份。中医学认为菝葜性味甘、微苦、涩、平,具有利湿去浊、祛风除痹、解毒散瘀的功效[1]。以菝葜药材为原料药的制剂在临床上应用广泛且疗效确切,如金刚藤软胶囊(国药准字Z20070006)[2],主治慢性盆腔炎、附件炎、子宫内膜炎等炎症性疾病[3]。目前,菝葜药材仍缺乏有效的质量控制方法,2015年版《中国药典》未收载其制剂。而在指纹图谱方面,周艳林等[4]分别对菝葜药材脂溶性成分和黄酮类成分运用 HPLC 指纹图谱进行了分析。杨立勇等[5]对贵州产菝葜及其混淆品光叶菝葜的指纹图谱进行了综合分析比较。徐贝等[6]则以湘产菝葜为研究对象进行HPLC指纹图谱分析。但金刚藤胶囊的原料药的质量控制还是很难推进。结合本课题组前期研究[7-8]和菝葜与抗盆腔炎大鼠谱效学研究[9]表明:菝葜药材的总黄酮成分是潜在的药效成分。为此,本研究采用HPLC法建立了菝葜药材指纹图谱,并对菝葜药材进行聚类分析,以期发现菝葜药材中总黄酮成分含量较高的产地,为其制剂原料药的质量控制提供实验依据。
1 仪器与材料
1.1 仪器
Ultimate 3000 DGLC高效液相色谱仪(美国Dionex公司,LPG-3400SD四元泵、TCC3000-RS柱温箱、WPS-3000SL自动进样器、DAD检测器、Chromeleon7.2数据处理软件);Agilent 1260 高效液相色谱仪(G1311B四元泵、G1316A柱温箱、G1315DDAD检测器、G1329B进样器);UFLC-Triple TOF-MS/MS超快速高效液相色谱串联四极杆飞行时间质谱仪(LC-20AD-XR二元泵,CTO-20A柱温箱,SIL-20AD-XR自动进样器,SPD-M20A PDA 检测器,日本岛津公司;Triple TOF 5600 plus,美国 AB SCIEX公司);中药粉碎机(DMF-8A,浙江温岭市铭大药材机械设备有限公司);超纯水器(Simplicity,美国Millipore公司);万分之一电子分析天平(ME204,瑞士Mettlertoledo公司);数控超声波清洗器(KQ500DE,昆山市超声仪器有限公司)。
1.2 材料
甲醇(分析纯,广州化学试剂厂,批号:2019010128);乙腈(色谱纯,Honeywell,批号:UN1648);磷酸(色谱纯,上海阿拉丁生化科技股份有限公司,批号:P112025);水为超纯水。
黄杞苷对照品(批号:11906-201102,纯度:93.7%)、槲皮苷对照品(批号:111538-201606,纯度:90.6%)(中国食品药品检定研究院);落新妇苷对照品(批号:A102725,纯度:98%)(上海阿拉丁生化科技股份有限公司);异黄杞苷对照品(批号:B24735,纯度:95%)(上海源叶生物科技有限公司)。
实验用20批菝葜药材均购买于广州市清平中药材专业市场,并经中山大学生命科学院廖文波教授鉴定为百合科植物菝葜(SmilaxchinaL.)的干燥根茎。药材来源见表1。
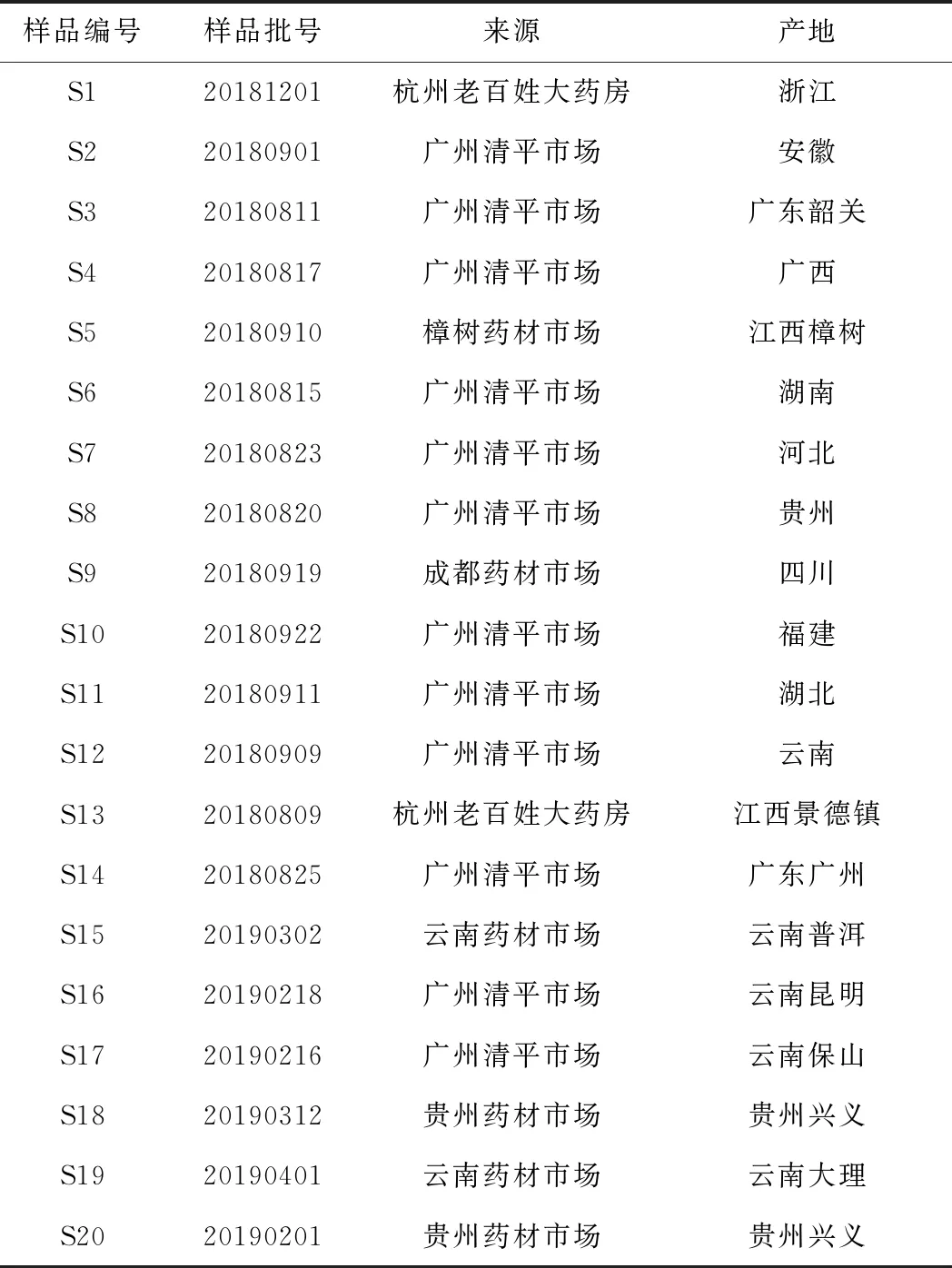
表1 20批菝葜药材来源信息
2 方法与结果
2.1 供试品的制备
精密称取菝葜药材粉末(过2号筛)0.5 g,置于锥形瓶中,精密加入甲醇25 mL,称定重量,超声(100 W,100 Hz)处理30 min,放冷,称重,用甲醇补足减失的重量。取上清液,用0.22 μm微孔滤膜过滤,续滤液作为供试品溶液。
2.2 对照品溶液的制备
精密称取落新妇苷、槲皮苷、黄杞苷和异黄杞苷对照品适量,加甲醇制成对照品储备液,浓度依次为122.6、55.0、502.4和85.0 μg/mL。
2.3 色谱条件
色谱柱:Hitachi High-Tech C18(4.6 mm×250 mm,5 μm),流动相:乙腈(A)-0.1%磷酸水溶液(B)梯度洗脱(0 min~18 min,10% A;18 min~120 min,10%~30% A),检测波长:303 nm,进样量:10 μL,体积流量:1.0 mL/min,柱温:30 ℃。理论板数按落新妇苷计算不得低于5000。采用《中药色谱指纹图谱相似度评价系统(2012版)》评价其相似度。
2.4 方法学考察
2.4.1 专属性试验
取“2.1”项下菝葜药材供试品溶液(样品编号:S3)、空白溶剂(甲醇)、对照品溶液,分别进样,用相同的色谱条件进行分析。结果表明专属性良好,见图1。

注:A:空白对照;B:混合对照品;C:菝葜供试品;8:落新妇苷;12:槲皮苷;13:黄杞苷;15:异黄杞苷。图1 空白对照、混合对照品和菝葜药材供试品的HPLC图谱
2.4.2 精密度试验
取同一批菝葜药材(样品编号:S3),精密称定,按“2.1”项下方法制备供试品溶液,按“2.3”项下色谱条件,连续进样6次,记录 HPLC 色谱图。根据对照品确认8、12、13、15号峰依次为落新妇苷、槲皮苷、黄杞苷和异黄杞苷。其中13号峰面积大,且稳定,设为参照峰,计算其他3个峰的相对保留时间与相对峰面积。结果显示:相对保留时间RSD在0.02%~0.08%之间,相对峰面积RSD在0.14%~2.05%之间,相似度均等于1.00。结果表明仪器的精密度良好。
2.4.3 稳定性试验
精密称取同一批菝葜药材(样品编号:S3),按“2.1”项下方法制备供试品溶液,按“2.3”项下色谱条件,分别在0、2、4、8、12、24、48、72、120 h 进样,记录 HPLC 色谱图。结果显示:相对保留时间RSD在0.02%~0.09%之间,相对峰面积RSD在0.25%~1.09%之间,相似度均等于1.00。结果表明供试品溶液放置120 h 内稳定。
2.4.4 重复性试验
取同一批菝葜药材(样品编号:S3),精密称定,按“2.1”项下方法分别制备6份供试品溶液,按“2.3”项下色谱条件分别进样,记录 HPLC 色图谱。结果显示:相对保留时间RSD在0.01%~0.04%之间,相对峰面积RSD在1.90%~3.54%之间,相似度均等于1.00。结果表明方法重复性好。
2.4.5 耐用性试验
取同一批菝葜药材(样品编号:S3),精密称定,按“2.1”项下方法制备供试品溶液,分别使用Hitachi High-Tech C18(4.6 mm×250 mm,5 μm,No.28H5I-019)、Thermo C18(4.6 mm×250 mm,5 μm,No.01705069)、SEPAX Amethyst C18(4.6 mm×250 mm,5 μm,S.N.11041698382)3种型号色谱柱,按“2.3”项下色谱条件进样,记录色谱图。结果显示:相对保留时间RSD在0.23%~0.64%之间,相对峰面积RSD在1.93%~3.54%之间,相似度均等于1.00,表明方法重复性好。
2.5 菝葜药材指纹图谱的构建
2.5.1 指纹图谱的获得
取表1中的菝葜药材按“2.1”项下方法处理,按“2.3”项下色谱条件分别进样,记录各色谱图。各批次样品叠加图结果见图2。
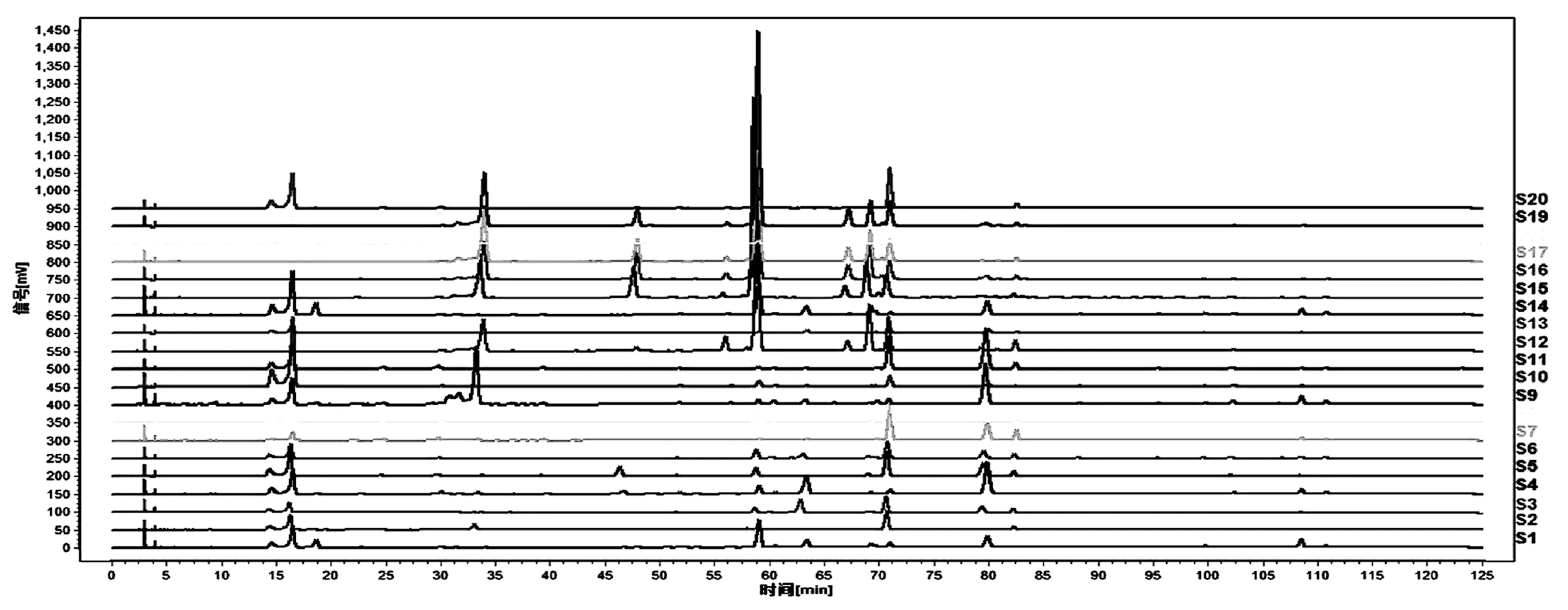
图2 20批菝葜药材HPLC指纹图谱
2.5.2 共有峰和参照峰的确认
分析20批菝葜样品的测定结果,获得19个共有峰(若某一图谱在某一保留时间处无峰,相应的峰面积为零,仍给相应的编号,以保证各图谱都有相同的色谱峰数),见图3和表3。因8号和13号峰稳定性好、分离度好且具有良好的抗炎活性[7,9],故选择该峰为参照峰。
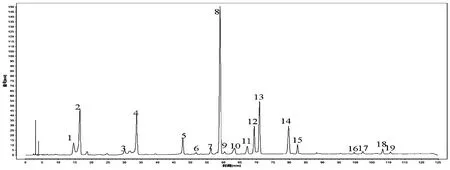
图3 不同批次菝葜药材指纹图谱共有模式
2.6 共有峰的确证
2.6.1 测定法
通过超快速高效液相色谱串联四极杆飞行时间质谱(UFLC-Triple TOF-MS/MS)技术对“2.1”及“2.2”项下菝葜供试品溶液(样品编号:S3)、对照品溶液、空白溶剂进行检测,色谱条件同“2.3”项下,分流进样。质谱条件:ESI电喷雾离子源,离子源喷雾电压:5 500 V,离子源气体压力:255 psi,离子源气体:155 psi,离子源温度:550 ℃,碰撞气压力:10 psi,气帘气压力:35 psi,扫描范围:m/z 50~2 000,采用正、负离子模式进行检测。
2.6.2 共有峰的确证
检测结果通过与碎片离子分析、保留时间、对照品对照及文献查阅[10-19],指认并确证菝葜药材指纹图谱中共有峰所对应的化合物。结果见表3。

表2 菝葜药材19个色谱峰的相对峰面积
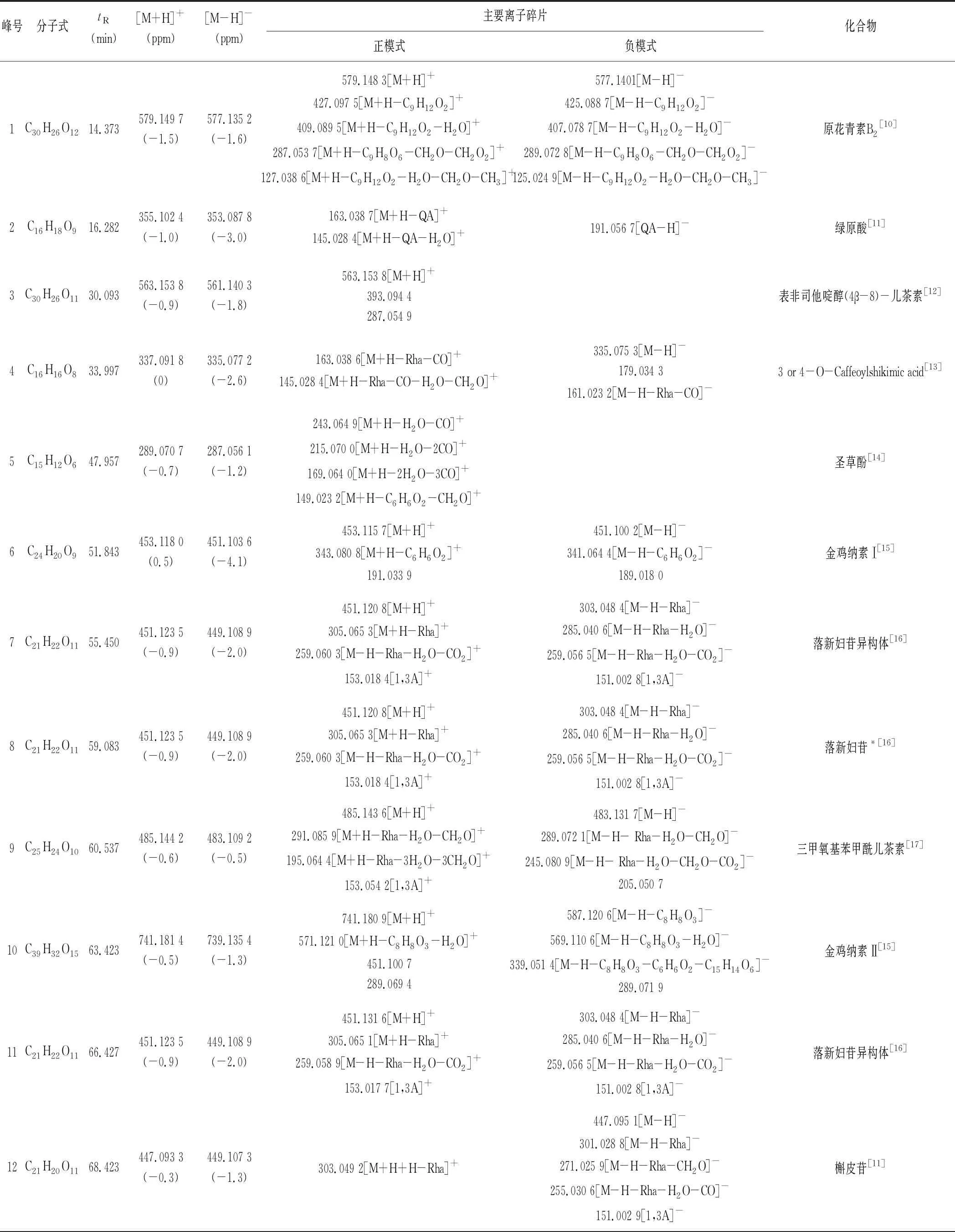
表3 菝葜药材的化学成分质谱归属

续表3
2.7 菝葜药材的聚类分析
采用IBM SPSS Statistics 24.0数据统计软件,将20批菝葜样品的19个共有峰的相对峰面积导入,使用瓦尔德聚类方法,用欧式平方距离计算样品相似度,聚类结果见图4。由聚类分析图可见:当欧式平方距离为8时,可将20批菝葜样品聚为3类。S12、S15、S16、S17、S19聚为第一类;S2、S5、S8、S10、S18、S20聚为第二类;S1、S3、S4、S6、S7、S9、S11、S13、S14聚为第三类。结合表2和表3可知,第一类的产地均为云南,且成分以8号峰落新妇苷为主,平均相对峰面积在48%左右;第二类的6批菝葜药材中1、2号峰(有机酸类)相对峰面积较高;第三类的9批菝葜药材中14、18、19号峰(芪类和皂苷类)相对峰面积较高。
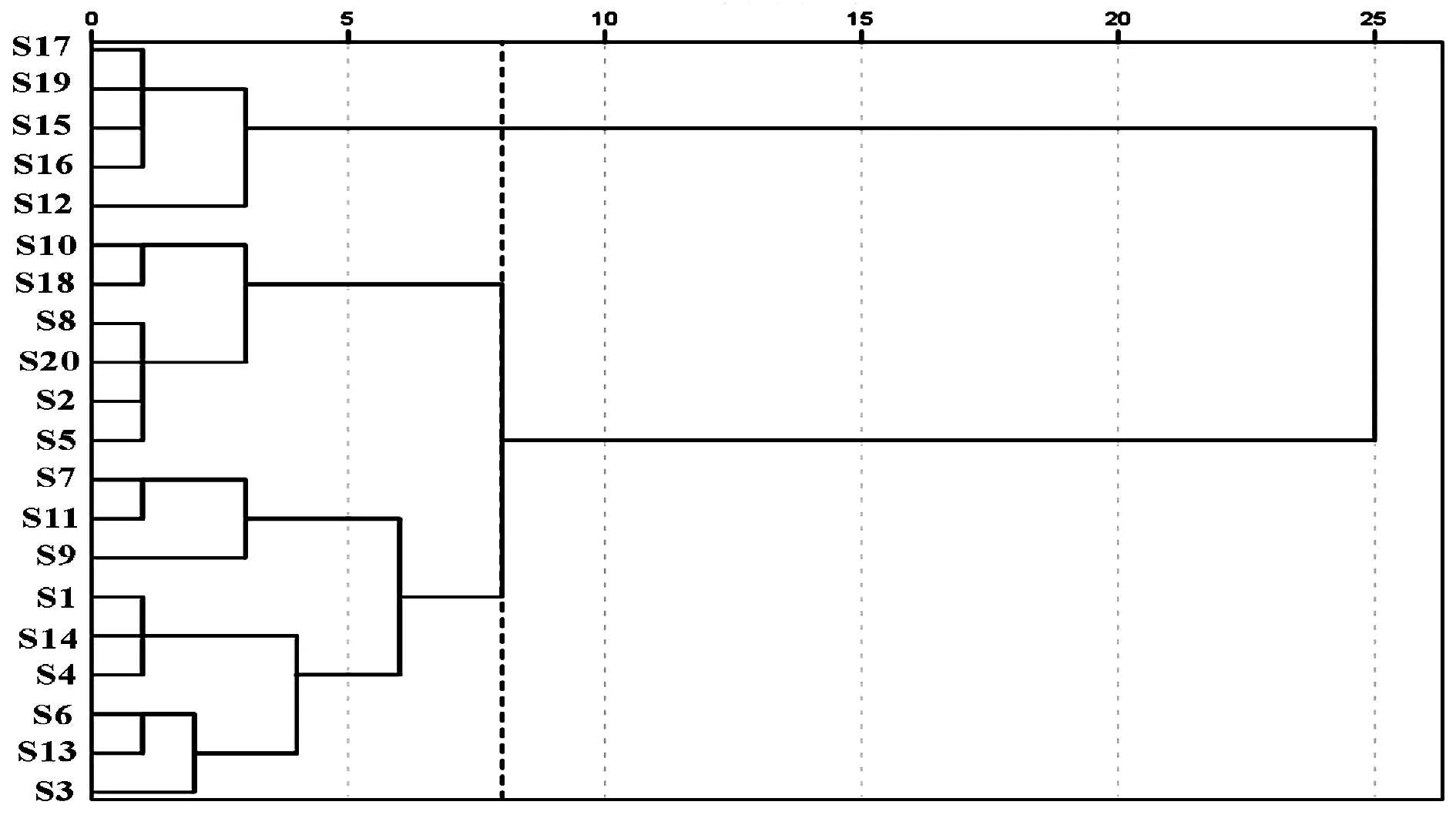
图4 20批菝葜样品的聚类分析图谱
3 讨论
中药指纹图谱是从中药成分群角度出发的一种客观、整体和多指标的质量评价方式,已被广泛应用于中药的质量控制评价[20-21]。本研究考察了柱温对指纹图谱的影响,发现30 ℃时各色谱峰形较好;采用DAD检测器考察了各色谱峰的紫外光谱对指纹图谱的影响,发现大部分化合物在290~320 nm处有较大吸收,故确定指纹图谱的检测波长为303 nm。
本研究构建了全国12个不同产地共20批次菝葜药材的HPLC指纹图谱,通过UFLC-Triple TOF-MS/MS技术指认了19个共有峰,并确证了其中17个成分(见表3),以有机酸类、黄酮类和皂苷类成分为主。因为药材产地来源的不同,在研究中发现各共有峰的相似度较小(<0.9),说明菝葜药材化合物成分含量受地理环境、气候影响较大。根据各产地菝葜药材聚类分析结果发现:云南产地的菝葜药材总黄酮化合物含量较高(落新妇苷、槲皮苷、黄杞苷和异黄杞苷总峰面积占比约为64.90%),且黄酮类化合物中以落新妇苷含量最高(平均峰面积约占48.53%)。而贵州产地的有机酸含量较高(平均相对峰面积约占44.93%)。这为菝葜制剂原生药材产地的筛选提供实验依据。
综上所述,此研究建立了菝葜药材HPLC指纹图谱分析方法。该方法准确度和精密度高,易于操作,适用性强。可为菝葜药材的质量控制评价提供新方法,也为菝葜药材的物质研究究奠定基础。
