CRISPR/Cas9介导的同源重组在非编码区疾病相关位点功能研究中的应用
2021-03-03侯国俊唐元家
徐 宁,周 甜,侯国俊,沈 南,唐元家
上海交通大学医学院附属仁济医院风湿病科,上海市风湿病学研究所,上海200127
全基因组关联研究(genome-wide association study,GWAS)报道了许多疾病相关的单核苷酸多态性(single nucleotide polymorphism,SNP) 位点[1-2],且进一步研究[3-4]发现这些位点大多位于具有调控活性的非编码区。由于技术手段的限制,对上述位点功能性的验证和机制的研究仅通过大样本相关性分析或双荧光素酶报告系统等较为简单又粗糙的方式进行,尚无法提供能够直接验证SNP功能的证据[5-6]。然而,规律成簇间隔短回文重复序 列/相 关 蛋 白9 [clustered regularly interspaced short palindromic repeats(CRISPR)/CRISPR associated protein 9,CRISPR/Cas9] 基因组编辑系统的出现改变了这一现状[7]。该系统由负责靶向基因组的单链导向RNA(single guide RNA,sgRNA) 与负责对基因组切割的Cas9 组成。在sgRNA 的作用下,Cas9 蛋白可诱导靶点DNA 序列处形成双链断裂(double-stranded break,DSB),随后经非同源末端连接(non-homologous end joining, NHEJ) 或 同 源 重 组 (homologous recombination,HR)修复途径分别产生位点敲除/插入或位点重组的细胞株,而这些细胞株则可为疾病易感位点的功能研究提供最直接的证据[8]。
系统性红斑狼疮(systemic lupus erythematosus,SLE)是一种复杂的自身免疫病。GWAS 报道显示多个SNP 位点均与SLE 的发生与发展密切相关,但大多数功能未明。因此,利用CRISPR/Cas9 基因组编辑系统对这些位点的功能性进行验证可直观地了解其与疾病的关系。研究[9-11]发现,Ⅰ型干扰素信号通路的异常活化是SLE的重要特征之一,且miR-146a 是干扰素信号通路强有力的负调控分子,在SLE 患者的单核细胞中异常低表达。GWAS 发 现SNP rs2431697 是SLE 的 易 感 位 点 之 一[12],表达数量性状位点(expression quantitative trait locus,eQTL)分析发现在欧洲人群中rs2431697 等位基因的变化会影响miR-146a 的表达[13]。故而推测,rs2431697 或其连锁位点可能存在对miR-146a 的调控作用[14]。借助HaploReg数据库(https://pubs.broadinstitute.org/mammals/haploreg/haploreg.php)对rs2431697 连锁位点进行分析后发现,SNP rs1978421 与之存在强连锁效应(r2=0.94)。而后,本课题组分别对rs2431697 与rs1978421 SNP 处的组蛋白修饰信号进行分析(数据源自ENCODE 数据库),结果显示在这2个位点处均探测到常见于转录调节活性区域的组蛋白H3 第4 位赖氨酸单甲基化(H3K4me1)和组蛋白H3 第27 位赖氨酸乙酰化(H3K27ac)修饰信号,故而认为这2 个位点都有可能位于具有转录调节活性的DNA 元件上,具备参与miR-146a 表达调控的能力。因此,本研究以rs1978421 为研究对象,采用CRISPR/Cas9介导的同源重组技术在单核细胞系U-937中研究该位点是否参与miR-146a 的表达调控,并以此来构建研究非编码区疾病相关SNP功能的有效方案。
1 材料与方法
1.1 实验材料
1.1.1 细胞株 HEK293T 细胞和U-937 细胞均购自中国科学院典型培养物保藏委员会细胞库。经鉴定,该2种细胞无支原体污染。
1.1.2 主要仪器与试剂 DMEM 培养基、RPMI-1640 培养基、胎牛血清(fetal bovine serum,FBS)以及Opti-MEM 无血清培养基(Gibco,美国),电转染系统NeonTMTransfection System、LipofectamineTM2000 转染试剂以及TRIzol 试剂(Invitrogen,美国),高速冷冻离心机(Eppendorf,德国),TaqManTMmiRNA 反转录试剂盒、miR-146a 与 对 照RNU48 定 量 探 针、TaqManTMmiRNA 定量试剂盒(Thermo,美国),PrimeScriptTMRT 试剂盒与TB Green®Premix Ex TaqTM试 剂 盒(TaKaRa,日本),ViiATM7荧光定量PCR系统(Applied Biosystems,美国),质粒pSpCas9(BB)-2A-GFP(#PX458;Addgene,美国),细胞株基因型鉴定试剂盒TransDirect®Animal Tissue PCR Kit(北京全式金生物技术有限公司),I-5TM2×High-Fidelity Master Mix (北京擎科生物科技有限公司),FACSAria Ⅱ流式分选仪(BD 公司,美国),血液/细胞/组织基因组DNA 提取试剂盒[天根生化科技(北京)有限公司]。
1.2 实验方法
1.2.1 细胞培养 U-937 细胞采用含10%FBS 的RPMI-1640 培养基进行培养,HEK293T 细胞采用含10%FBS 的DMEM 培养基进行培养。培养条件为37 ℃、5%CO2。本实验所用细胞均处于指数生长期,状态良好。
1.2.2 U-937 野生型rs1978421 位点基因型鉴定 为获得靶细胞rs1978421 位点等位基因同源重组的细胞系,首先需对U-937 细胞rs1978421 位点处的碱基类型进行鉴定(该基因型为野生型)。取约2×105个U-937 细胞,离心后弃上清液,使用血液/细胞/组织基因组DNA提取试剂盒提取基因组DNA。取100 ng 基因组DNA 进行rs1978421 位点的PCR 扩增,扩增反应采用I-5TM2×High-Fidelity Master Mix 高保真酶反应液进行,反应体系(50 μL)如下:模板DNA 100 ng、引物(10 μmol/L)各2.5 μL、2×高保真酶反应液25 μL。将各成分混合均匀后短时离心,按照说明书中的条件进行PCR反应。引物序列见表1,产物长度500 bp。当反应结束后,通过测序公司进行取样并测序。
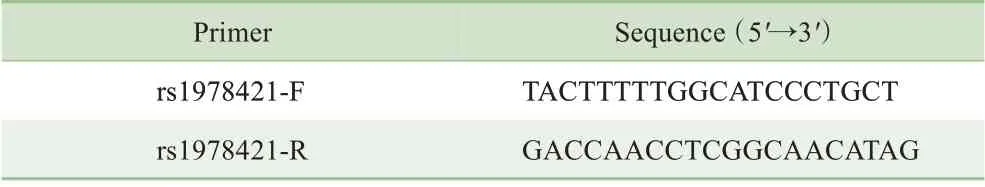
表1 rs1978421位点鉴定引物Tab 1 Indentification primer of rs1978421 locus
1.2.3 sgRNA/Cas9 表达载体的构建 选择pSpCas9(BB)-2A-GFP为sgRNA/Cas9的表达载体。将其进行酶切并电泳、回收后,与sgRNA 片段连接。利用IDT 在线sgRNA 设 计 工 具 (https://sg. idtdna. com/site/order/designtool/index/CRISPR_CUSTOM)对sgRNA 进行设计,原则如下:①SNP 位点在sgRNA 序列中。②sgRNA 靶向评分(on-target score)较高、脱靶评分(off-target score)较低。基于该原则,我们选取了靶向评分为71 分、脱靶评分为0 分的sgRNA,并以单链的形式合成(序列见表2)。将合成的sgRNA 正链与负链在体外复性为双链后,与上述酶切载体于4 ℃连接过夜,而后行转化实验,次日挑取单菌落进行测序。对测序成功的菌株抽提px458-rs1978421质粒备用。

表2 sgRNA功能序列及单链引物Tab 2 sgRNA functional sequence and single strand primers
1.2.4 质粒转染 于转染前1 d 向24 孔板加入HEK293T细胞,每孔2×105个,过夜培养后即可行转染实验。向每孔添加LipofectamineTM2000 转染试剂2 μL,其余操作均参照说明书进行。当转染结束后,继续培养48 h 即可行后续实验。将U-937细胞离心去培养基后,用磷酸盐缓冲液洗涤2 次,计数后进行分装,即每1.5 mL 离心管中加入1.5×106个细胞。离心后弃上清液并用电转缓冲液重悬,加入10 μg px458-rs1978421质粒(1 μg/μL)以及30 μg单链DNA 重组模板(序列见表3),电转条件为1 400 mV、10 ms、3 pulse。在电转后的第12 h加入DNA 连接酶Ⅳ的抑制剂Scr7(终浓度为1 μmol/L),以抑制NHEJ 途径对细胞双链DNA断裂的修复。
1.2.5 T7核酸内切酶Ⅰ实验 收集经px458-rs1978421质粒转染48 h 后的HEK293T 细胞,提取基因组DNA 后经PCR 反应对包含SNP 位点的片段进行扩增。取经纯化后的产物300 ng 进行复性,反应体系为15 μL,包含1.5 μL T7 核酸内切酶Ⅰ(T7 endonuclease Ⅰ,T7EⅠ)缓冲液;反应条件如下:95 ℃保持10 min,后以2 ℃/s 的速度降至85 ℃,再以0.3 ℃/s 的速度降至25 ℃,以25 ℃保持10 s后正常降温至4 ℃。待反应结束后,向每管复性产物加入2 μL T7EⅠ,于37 ℃孵育1 h 后用2%琼脂糖凝胶电泳检测切割效率。切割效率计算公式如下:

表3 同源重组模板序列Tab 3 Sequence of homologous recombination template

其中,I500bp、I310bp和I190bp分别为500 bp、310 bp 和190 bp条带的荧光强度值。
1.2.6 RNA 抽 提 与qPCR 检 测 使 用TRIzol 法 提 取RNA,以三氯甲烷萃取TRIzol 中的水相,并以异丙醇沉淀其中的RNA,用75%乙醇洗涤沉淀2 次后室温晾干。待RNA 充分干燥后,用20 μL 焦碳酸二乙酯(diethyl pyrocarbonate,DEPC)处理水进行溶解并测定浓度,将其浓度统一调至50 ng/μL 后利用TaqManTMmiRNA 反转录试剂盒进行miRNA qPCR 模板的制备,并将制备好的模板稀释3 倍后再行定量。利用PrimeScriptTMRT 反转录试剂盒将提取的总RNA 中的mRNA 反转录为cDNA,稀释6 倍后作为qPCR 的模板。利用TB Green®Premix Ex TaqTM试剂盒对稀释后的cDNA 模板进行qPCR 检测。结果以相对表达量2-ΔΔCT展示。qPCR引物序列见表4。
1.2.7 细胞分选 U-937 细胞转染48 h 后即可进行流式分选。以未转染的U-937细胞作为阴性对照来调节电压及相关参数;将活细胞与单个细胞依次设门圈出,通过调节异硫氰酸荧光素(fluorescein isothiocyanate,FITC)通道的电压将阴性对照的细胞集中在横坐标103左侧,即可将转染过的U-937 细胞上样;分选FITC 荧光通道横坐标103右侧的FITC 阳性的U-937 细胞群至96 孔细胞培养板,每孔1 个细胞。分选结束后于37 ℃、5%CO2培养箱中培养约14 d后即可进行基因型鉴定实验。
1.2.8 细胞基因型鉴定 在分选后的第14 日,96 孔板底部已有肉眼可见的细胞群落。使用移液器将细胞群落均匀地吹打成细胞悬液,吸取5~10 μL至96孔PCR反应板中,用来制备鉴定基因型的PCR 模板,剩余细胞悬液转至24 孔板中继续培养。模板制备采用TransDirect®Animal Tissue PCR 试剂盒,反应体系为50 μL,包括4~8 μL 模板。其余实验步骤按照试剂盒说明书进行。

表4 qPCR引物序列Tab 4 qPCR primers
1.3 统计学分析
应用GraphPad Prism 7.0 软件对qPCR 数据进行分析。通过非配对t检验比较野生型与重组型细胞中的对应基因表达的差异。P<0.05表示差异具有统计学意义。
2 结果
2.1 rs1978421位点sgRNA设计及效率鉴定
利用CRISPR/Cas9 基因组编辑系统研究非编码区SNP 位点的功能,具体流程如图1A。为设计针对rs1978421 位点进行同源重组的sgRNA,我们首先对U-937 细胞rs1978421 位点处的碱基类型进行鉴定;结果(图1B)发现在U-937 和HEK293T 细胞内,rs1978421 位点的等位基因均为胸腺嘧啶(T)。之后,我们以含有T等位基因的DNA 片段进行sgRNA 设计及载体px458-rs1978421 的构建(图1C)。成功获得载体后,我们在HEK293T 细胞中对sgRNA 切割效率进行检测。T7EⅠ核酸内切酶切割实验结果(图1D)显示,目的sgRNA 在HEK293T中rs1978421位点的切割效率约为24%,表明该sgRNA 能够对rs1978421 位点进行有效切割,可以应用于同源重组实验。
2.2 同源重组细胞株的构建与鉴定
鉴于rs1978421 可能与miR-146a 的表达相关,而miR-146a 在HEK293T 中低表达,在U-937 中高表达,因此我们选择在U-937细胞中进行同源重组实验。将载体以及同源重组模板通过电穿孔方法转入U-937 细胞48 h 后,通过流式分选仪将单个绿色荧光蛋白(green fluorescent protein,GFP)强阳性的细胞分选至96 孔板中,确保每孔只含有1 个细胞。在进行流式分选时发现,活细胞中GFP 阳性的细胞数占总数的2.29%(图2A)。经分选共得到960 个只含有1 个GFP 阳性U-937 细胞的培养孔。经2周培养后,有141个细胞增殖至肉眼可见的细胞群落;将rs1978421 位点附近的基因组DNA 经PCR 扩增并测序发现,有86 个细胞克隆基因为杂合子,占总数的61.0%;有25个细胞基因组未发生改变,占总数的17.7%;有9个细胞在rs1978421位点旁侧检测到不明来源DNA序列的插入,占总数6.4%;有15个细胞在rs1978421旁侧发生不同数目碱基删除,占总数的10.6%;另有6个细胞rs1978421位点的碱基成功由胸腺嘧啶(T)转变为胞嘧啶(C),重组成功细胞数占总数的4.3%(图2B、C和表5)。
2.3 rs1978421不同等位基因对周围基因表达影响
rs1978421 位于5 号染色体PTTG1 基因下游约27.4×103bp 以及miR-146a 的初级转录本miR3142HG 上游约12.1×103bp 处,其邻近2 Mbp 区域内的基因包括SLU7、TTC1、PWWP2A等(图3A)。为了检测rs1978421位点的调节基因,我们首先采用qPCR 法检测miR-146a 在不同基因型细胞中的表达水平;结果(图3B)显示,与TT基因型相比,在CC 型、SNP 位点邻近序列切除8 bp 以及SNP 位点邻近序列插入65 bp 的细胞克隆中,miR-146a 表达无差异。同时,对rs1978421 邻近的高表达基因检测表明,不同基因型对这些基因的表达也无显著影响(图3C)。基于以上数据,我们认为rs1978421 位点可能不是一个具有功能性的调节位点。

图1 非编码区SNP位点研究策略及rs1978421位点sgRNA设计及切割效率检测Fig 1 Research strategy for SNP located in non-coding region and rs1978421 sgRNA design and cutting efficiency test

图2 流式分选与细胞基因型鉴定Fig 2 FACS sorting and cell clones genotype identification

表5 SNP rs1978421同源重组实验所得基因型细胞株汇总Tab 5 Summary of genotypes of cell lines obtained in SNP rs1978421 homologous recombination
3 讨论
CRISPR/Cas9基因组编辑系统是一种可靠的非编码区疾病相关性SNP 功能研究手段。得益于该研究方法,许多疾病相关的功能性位点参与基因表达调控的方式被报道:如SNP rs1421085 等位基因的改变可加剧肥胖[15],SNP rs11672691 等位基因的变化与前列腺癌细胞的增殖相关[16]。在本研究中,利用CRISPR/Cas9 基因组编辑系统介导的同源重组成功得到了rs1978421 等位基因重组的U-937 细胞克隆,通过荧光定量PCR 检测rs1978421 周围基因的表达变化并以此明确了rs1978421 位点不是功能性的疾病易感位点。经过对rs1978421 和rs2431697 位点遗传学数据的进一步挖掘和分析,我们推测后者可能是参与miR-146a 的表达调控的功能性位点。后续我们将进一步对这个位点进行功能性鉴定。
本研究在SNP 位点的选择时仅以H3K4me1 与H3K27ac这2个组蛋白修饰信号作为依据,并未对SNP位点处的信息学数据进行深入挖掘。由于同源重组技术工作量大且实验周期长,因此对位点的选择尤为重要。在位点选择时,不仅要分析SNP 位点所在区域的组蛋白修饰情况,还要从其他角度进行信息挖掘:如参考ENCODE 数据库中转录因子Chip-seq 的数据对SNP 位点所在区域转录因子的结合情况进行分析,或是根据Haploreg 数据库对SNP 位点处转录因子结合位点进行预测以及分析SNP 等位基因的变化是否会影响转录因子结合等信息。由于增强子对转录调控不受基因组上距离的限制,因此在鉴定SNP 的功能性时,最好的办法是对不同等位基因细胞株进行全转录组测序,通过比较两者存在差异表达的基因来寻找SNP潜在的靶基因。
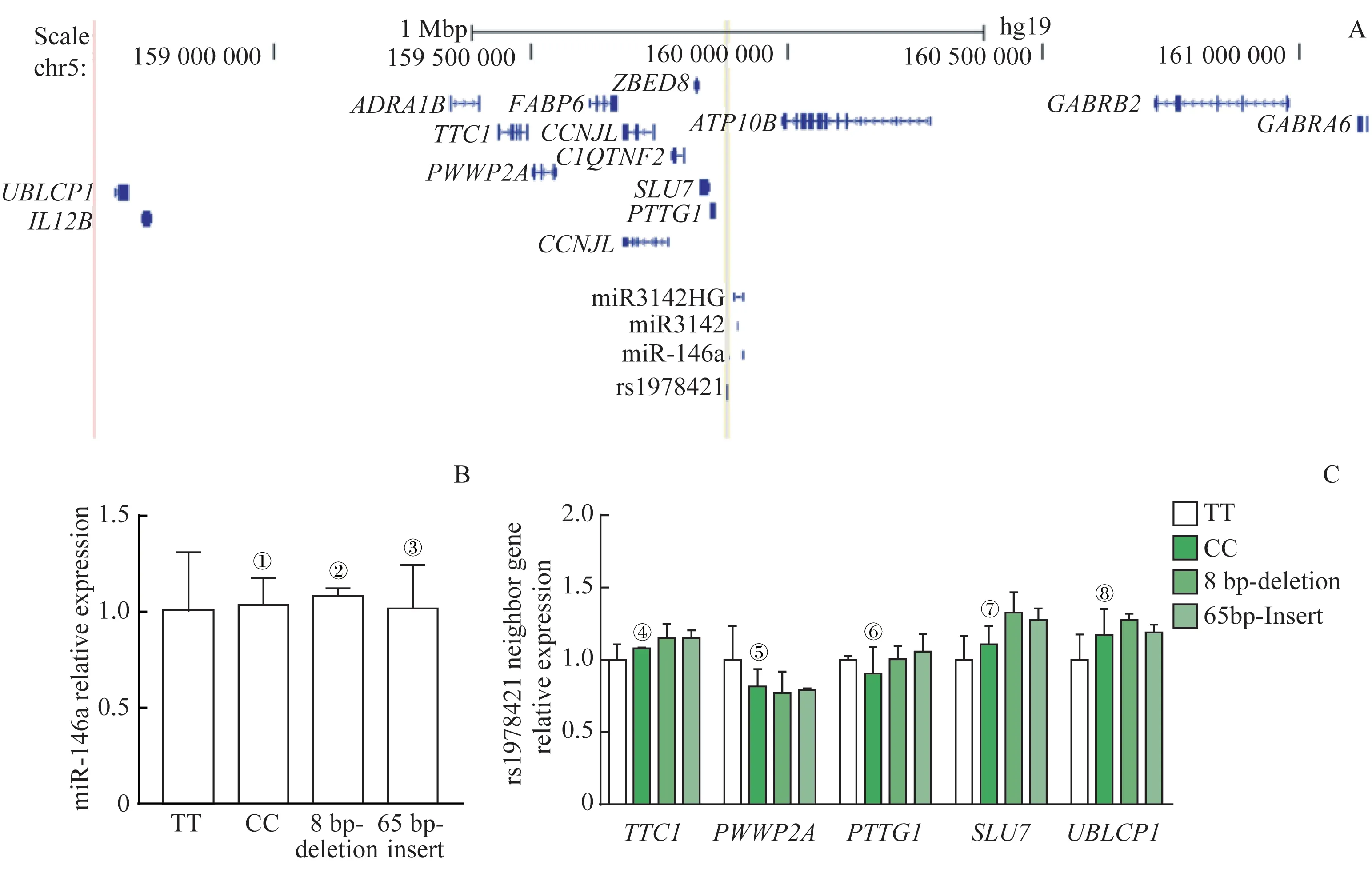
图3 不同基因型细胞中rs1978421邻近基因表达分析Fig 3 rs1978421 neighbor genes expressions analysis in cell clones with different genotypes
CRISPR/Cas9基因组编辑系统及其衍生工具系统的出现极大地推动了基因组非编码区调控功能的研究,如CRISPR 激活(CRISPR activation,CRISPRa)系统可对新的调控元件进行鉴定[17],CRISPR 干扰(CRISPR interference,CRISPRi) 系统可特异性地抑制基因表达[18]。由于这些转录调控的系统均依赖于sgRNA 与基因组DNA 序列的碱基互补配对,因此我们设想了一种等位碱基特异性的表达调控系统:已知sgRNA 与基因组DNA之间的碱基错配会影响两者之间互补配对效率[19-20],那么如果能够证明rs2431697 对miR-146a 存在转录调控作用,则可以把该区域作为靶点,利用CRISPR激活系统上调miR-146a 的表达,以增强对过度活化的干扰素信号通路的负调控作用。sgRNA 设计策略如下:通过分析SLE患者基因组样本确定rs2431697 位点在患者中的高频碱基,以携带高频碱基的DNA 片段为模板设计包含SNP 位点的sgRNA序列,在含有rs2431697不同等位碱基的细胞系中验证sgRNA 与DNA 的结合效率,筛选与携带高频等位碱基的基因组DNA 结合能力强而与携带低频等位碱基的基因组DNA结合能力弱的sgRNA。而此sgRNA设计策略使得该基因表达激活系统仅在携带SNP 风险等位基因的患者体内发挥效应,对携带正常等位基因的患者则表现为低干预效应或无干预效应,以减少对其免疫稳态的影响[21],从而建立了一种致病等位基因特异性的干预策略。
总之,环境因素和遗传因素共同参与了诸多疾病的发展。虽然这些疾病发生的机制仍未明确,但随着疾病易感基因和相关位点的功能机制研究日趋深入,人们对疾病的理解也将会更加全面。
