肝细胞癌发生发展关键基因及其功能的生物信息学分析
2021-02-27李文菠孙成杰周国俊应伟冯彦超黄婷侍琳黄理政李健水冷政伟
李文菠,孙成杰,周国俊,应伟,冯彦超,黄婷,侍琳,黄理政,李健水,冷政伟
(川北医学院附属医院 1.肝胆外科二 2.肿瘤干细胞研究中心,四川 南充 637000)
肝细胞癌(hepatocellular carcinoma,HCC)是消化系统最常见的恶性肿瘤之一,据美国癌症协会统计,HCC在男性癌症中的发病率排第六位,但在女性中仍以每年超过2%的速度增长,其病死率在男性排第五位,在女性中排第六位[1-2],与其他癌症相似,HCC与潜在的危险因素相关,如超重、吸烟、酗酒、乙型肝炎病毒(HBV)、丙型肝炎病毒(HCV)、非酒精性脂肪性肝病和某些基因突变等[3-4]。目前HCC的常用治疗方法包括手术切除、射频消融、肝脏移植、放射栓塞、免疫治疗等[5-7],不同分期的肿瘤患者选择不同的治疗方法,但5年复发率仍然很高[8-9]。近年来越来越多的研究表明,基因失活和基因突变是导致HCC发生发展的重要因素,故准确找到导致HCC的相关基因,从基因水平上研究HCC已成为研究热点之一。
近年来利用生物信息学和基因芯片技术研究因基因失活或基因突变所致肿瘤的发生、发展已成为发展趋势[10]。基因芯片具有数据全面,样本量大等优点,在生物学及医学领域占据重要位置。但这些数据信息混杂、缺乏足够实验基础,其准确性需要进一步验证。因此如何挖掘出准确可靠的基因信息逐渐成为生物信息学研究热点。本研究通过生物信息学的方法对HCC和癌旁组织的基因芯片中的差异基因进行分析并进行临床样本表达验证,筛选出与HCC发生发展的关键基因并进行临床样本验证,以期为其早期诊断、靶向治疗等提供参考依据。
1 资料与方法
1.1 芯片数据信息
HCC芯片从NCBI-GEO(https://www.ncbi.nlm.nih.gov/geo)数据库下载,GSE14520,GSE41804,GSE45267分别包含HCC组织225、20、48例,癌旁组织220、20、39例。
1.2 差异表达基因(differentially expressed genes,DEGs)筛选
通过GEO2R在线工具,定义HCC和癌旁组织,比较出DEGs,再筛选出满足绝对值LogFC>2且校正P<0.05的DEGs,将筛选出LogFC>2的基因,定义为上调DEGs,LogFC<-2的基因,定义为下调DEGs,通过Venn(http://bioinformatics.psb.ugent.be/webtools/Venn)在线制图工具筛取出3个芯片的上调基因的交集与下调调基因的交集。
1.3 GO 功能分析和KEGG 通路富集分析
利用生物学信息注释数据库(Database for Annotation,Visualization and Integrated Discovery,DAVID)为DEGs进行系统综合的生物学功能注释分析。通过上传DEGs到DAVID网站(https://david.ncifcrf.gov),进行GO(gene ontology,GO)功能分析和KEGG(Kyoto Encyclopedia of Genes and Genomes)通路富集分析,以P<0.05且FDR<0.05为差异有统计学意义,选择分析项目为生物过程(biological process,BP)、分子功能(molecular function,MF)、细胞成分(cellular component,CC)和信号通路。
1.4 蛋白相互作用网络PPI 分析和核心DEGs 筛选
将所获得的共同表达DEGs用STRING网站(https://string-db.org)进行PPI网络图的构建,构建后的网络图利用Cytoscape3.7.2软件及MCODE插件分析,筛选出核心DEGs。
1.5 核心DEGs 生存分析
在Kaplan-Meier Plotter网站(http://www.kmplot.com)中,将核心DEGs逐个进行生存分析,选择总生存率(OS)为指标,作出每个基因的生存曲线图,筛选出生存分析中P<0.05的基因,其可能为与预后相关基因。
1.6 预后相关DEGs 在HCC 与癌旁组织中的表达情况
把Kaplan-Meier Plotter网站中筛选出的与预后相关的基因用GEPIA(http://gepia.cancer-pku.cn)在线分析,选择表达图形选择Box Plots,数据来源选择LIHC,得到其在HCC组织和癌旁组织中的表达情况,以P<0.05为具有统计学意义。
1.7 与预后相关且在HCC 中高表达基因的功能及通路富集分析
将获得的与预后相关且在HCC中高表达基因上传至Metascape网站(http://metascape.org/gp/index.html#/main/step1),种属选择homosapiens,得到功能和通路的富集分析结果,以P<0.05为具有统计学意义。
1.8 免疫组化染色分析
在本研究中心选取HCC组织和癌旁组织各70例用于将石蜡包埋的组织切成5μm的切片。使用标准的免疫过氧化物酶染色程序进行免疫组化分析。分析染色强度(阴性:0分,弱阳性:1分,中等阳性:2分,强阳性:3分)和阳性细胞百分比(<5%:0分,5%~25%:1分,26%~50%:2分,51%~75%:3分,76%~100%:4分),最终计算公式:强度×百分比,其最终范围为0~12分。用这种方法对每一张玻片进行打分,以6分为界限,分为低表达样本和高表达样本。然后使用SPSS分别对HCC组织和癌旁组织进行数据统计,再把统计数据导入GraphPad prism 8中绘制统计图。
2 结 果
2.1 DEGs 筛选结果
芯片GSE14520,GSE41804,GSE45267分别筛选出差DEGs 252、257、497个,其中上调的DEGs分别为54、63、116个,下调的DEGs分别为198、194、381个,3个芯片的共差异的上调基因为16个(图1A),共差异的下调基因为62个(图1B),其共同的DEGs具体详见(表1)。
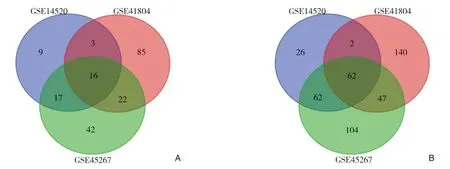
图1 三个芯片DEGs 的Venn 图(上调基因LogFc>2,下调基因LogFc<-2) A:16个上调DEGs;B:62个下调DEGsFigure1 Venn diagram of DEGs in the three gene microarrays (up-regulated LogFc>2,down-regulated LogFc<-2) A:16 upregulated genes;B:62 down-regulated genes

表1 78个共同DEGsTable1 The 78 common DEGs
2.2 GO与KEGG分析结果
经过DAVID网站将差异表达的78个基因,进行GO功能分析和KEGG通路富集分析。
GO功能分析中(表2),BP共涉及48个方面,主要集中在:细胞负增长的调控、细胞对锌离子的反应、外源性药物代谢过程、异型生物质的代谢过程、氧化还原过程、细胞对镉离子反应、P450表氧化酶通路、类固醇代谢过程;MF共有21个相关方面,其中主要集中于氧化还原酶活性、铁离子结合、氧结合、血红素结合、单加氧酶活性、花生四烯酸环氧合酶活性、氧化还原酶活性、类固醇羟化酶活性、咖啡因氧化酶活性、芳香酶活性;CC共涉及13个方面,其中有统计学意义的有以下方面:细胞器膜、细胞外泌体、细胞外区、内质网膜、细胞外间隙、胞质核周区、中间体、血液微粒、次膜攻击复合物。KEGG通路富集分析(表3)中,共涉及15个通路,其主要涉及于视黄醇的新陈代谢、矿物质吸收、药物代谢-细胞色素P450、化学致癌性、细胞色素P450对外源生物的代谢作用、咖啡因代谢、甾体类激素生物合成、亚油酸代谢、代谢途径、P53信号通路、药物代谢-其他酶。
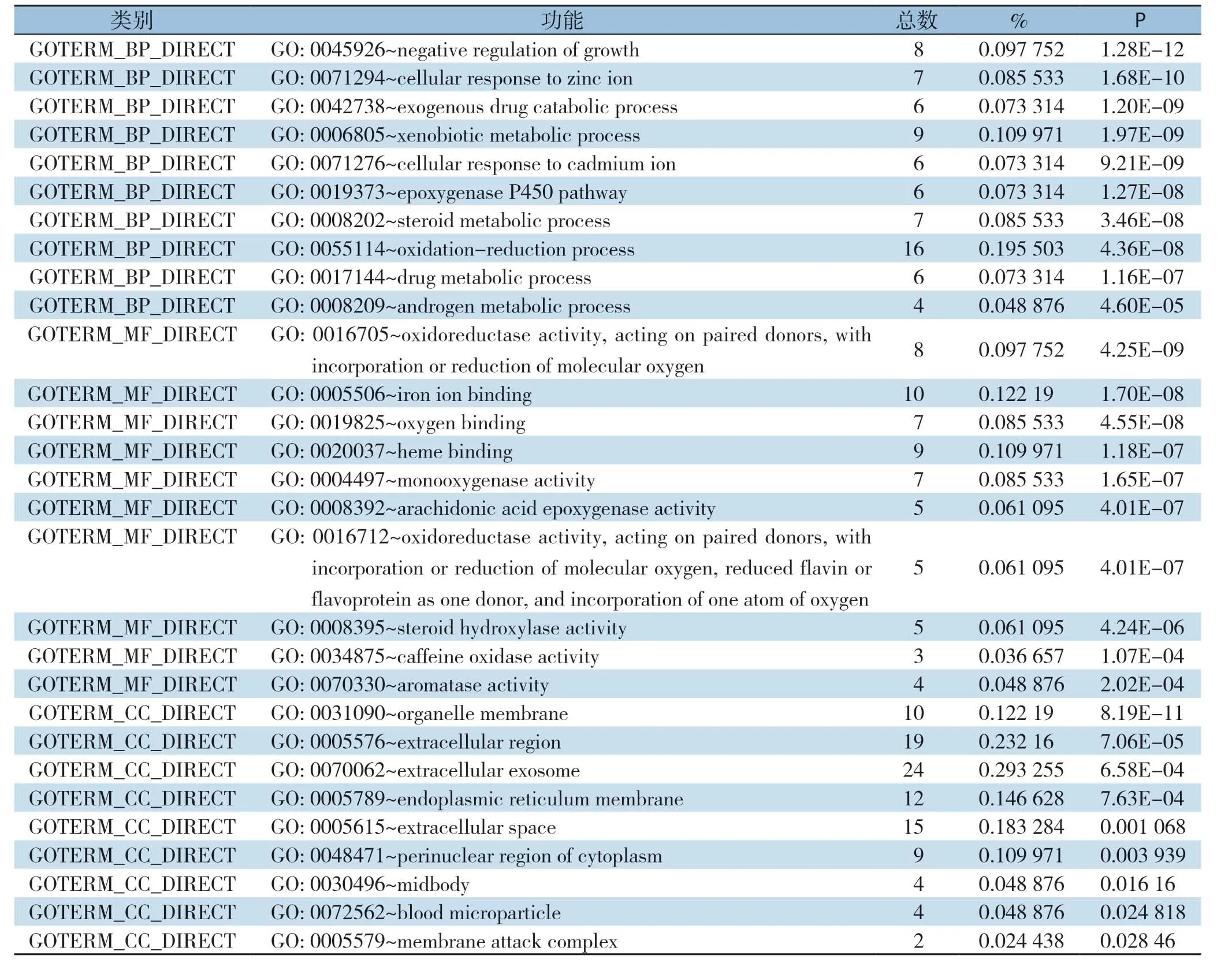
表2 78个DEGs GO 功能分析Table2 GO function analysis of the 78 DEGs

表3 78个DEGs 的KEGG 通路富集分Table3 KEGG pathway enrichment analysis of the 78 DEGs
2.3 PPI分析结果
将78个DEGs基因上传至STRING网站分析后,有15个基因未出现在PPI网络分析中,剩余共有63个基因,其中包括上调基因17个,下调基因46个,共有蛋白之间相互作用关系线条156条(图2A)。通过Cytoscape 3.7.2软件及MCODE插件分析后,得到两簇相交点最多的基因簇,分别有9、8个基因,分别有36、21条线,其分值分别为9、6,将其定义为核心EGs。共获得22个核心DEGs(图2B)。
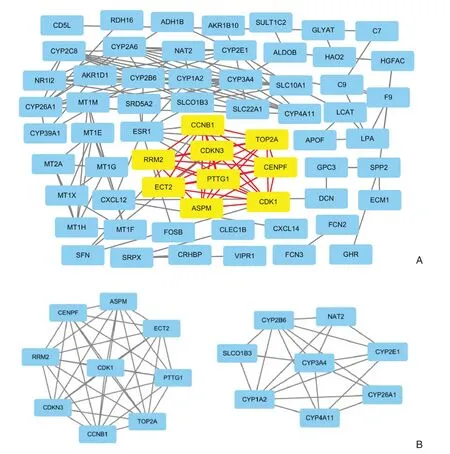
图2 共同DEGs 的PPI分析(黄色表示上调基因,蓝色表示下调基因) A:63个DEGs 的PPI 图;B:22个核心共同DEGsFigure2 PPI analysis of the common DEGs (yellow color representing up-regulated genes,blue color representing down-regulated genes) A:PPI network of the 63 DEGs;B:The 22 common core DEGs
2.4 核心DEGs 生存分析结果
17个核心DEGs经过Kaplan-Meier Plotter网站生存分析后,共有9个基因(CDK1、ASPM、CENPF、RRM2、CCNB1、TOP2A、PTTG1、ECT2、CDKN3)的生存分析差异有统计学意义(均P<0.05),另8个基因的生存分析差异无统计学意义(均P>0.05)(图3)。
2.5 预后相关DEGs表达量分析结果
将上述9个与预后相关的DEGs在GEPIA网站进行表达量分析后,9个基因均在HCC组织中较癌旁组织高表达(均P<0.05)(图4)。
2.6 高表达核心DEGs功能及通路富集分析结果
将上述9个在HCC组织中高表达的基因上传至Metascape网站分析,得到功能和通路的富集分析主要集中表现在细胞有丝分裂的负调控、细胞周期、核染色体隔离和雌配子的产生等方面(图5)(表4)。
2.7 关键DEGs验证结果
在筛选出来的9个基因中选取CDK1在HCC组织和癌旁组织中的染色,结果显示,CDK1在HCC组织中的评分为(7.871 8±1.524 9)分,在癌旁组织中的评分为(3.410 3±1.163 4)分,差异有统计学意义(t=14.429,P<0.0001)(图6)。
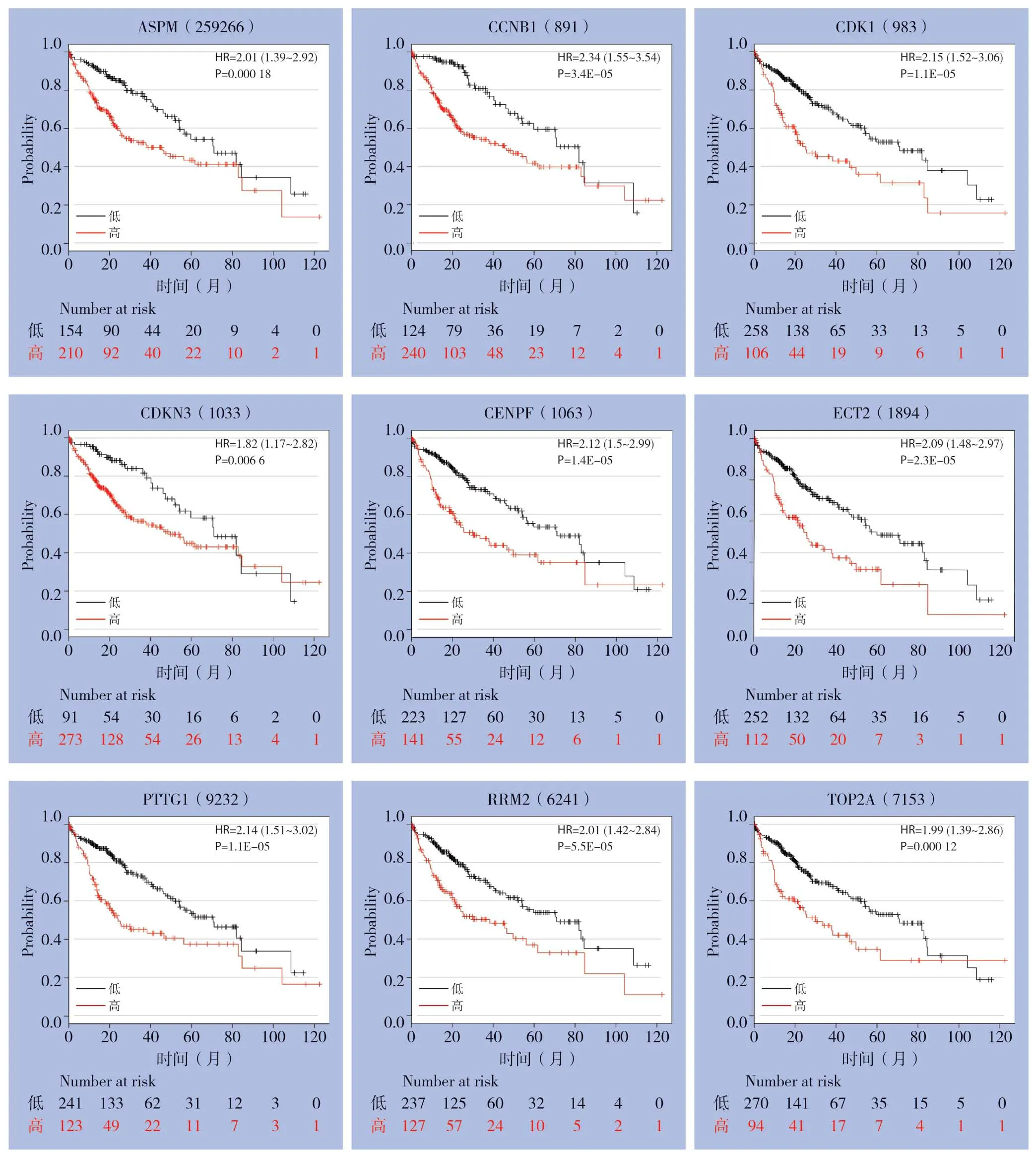
图3 核心DEGs 生存分析图Figure3 Survival curves of the core DEGs
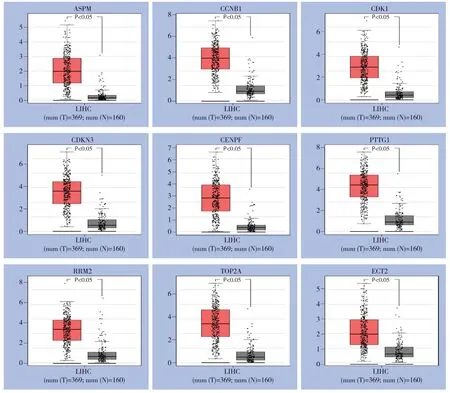
图4 9个DEGs 在组织中表达盒形图Figure4 Box plots of expressions of the 9 DEGs
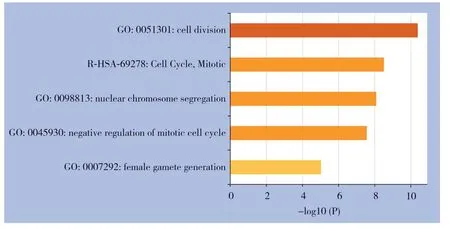
图5 9个核心DEGs 功能和通路富集图Figure5 Function and pathway enrichment plots of the 9 core DEGs
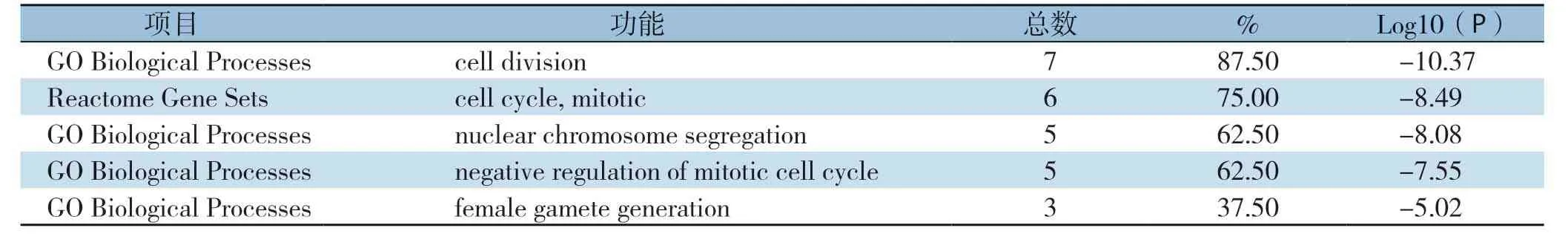
表4 9个核心DEGs 功能和通路富集分析数据Table4 Function and pathway enrichment data of the 9 core DEGs

图6 免疫组化检测CDK1 在HCC 与癌旁组织的表达Figure6 Immunohistochemical staining for CDK1 expressions in HCC and adjacent tissue
3 讨 论
HCC的发生发展通常是涉及基因、环境、饮食等多种因素的共同作用过程,利用生物信息学技术准确筛选出导致HCC发生发展的关键基因,这对于HCC的早期诊断、精准靶向治疗提供了重要依据。近年来伴随着生物信息学的蓬勃发展,大量基因芯片应用于研究疾病的发生发展和靶向基因的筛选等方面。例如Cao等[11]利用3个GEO数据库进行生物信息学分析,确定参与IL-10信号转导的CXCL8、CXCL1和IL-1β是溃疡性结肠炎的前3个核心基因;Mo等[12]利用生物信息学分析确定DNAJB4作为潜在的乳腺癌标记物;Xue等[13]综合生物信息学分析确定了4个(CDC45、GINS2、MCM2和PCNA)可能与宫颈癌患者预后相关的关键基因,可作为宫颈癌潜在的预后生物标志物。本研究基于生物信息学的分析方法在GEO数据库中筛选出近几年的、样本量较大的3份HCC及癌旁组织基因芯片,并在多个生物信息分析网站中进行了系统全面的分析,最终得出CDK1(细胞周期蛋白依赖性激酶1)、ASPM(纺锤体微管组装因子)、RRM2(核糖核苷酸还原酶调节亚基M2)、TOP2A(DNA拓扑异构酶2A)、CENPF(着丝粒蛋白F)、CCNB1(细胞周期蛋白B1)、PTTG1(垂体肿瘤转化基因1)、ECT2(上皮细胞转化序列2)、CDKN3(细胞周期蛋白依赖性激酶抑制因子3)与HCC发生、发展有重要关系,并最后在本研究中心选取临床样本进行表达验证。筛选出的9个基因主要作用于细胞有丝分裂的负调控、细胞周期、核染色体隔离和雌配子的产生等方面,从而引起细胞周期的紊乱、基因的突变,最终导致癌症的发生、发展。
CDK1属于丝氨酸/苏氨酸蛋白激酶家族,是调节细胞周期进程、DNA复制和分离、细胞成熟和增殖所必需的,CDK1的异常激活通过促进细胞增殖在肿瘤发生中起重要作用。CDK1在多种癌症中均有表达,在结直肠癌中,CDK1作为miR-769的直接靶点,在癌组织中高表达,miR-769通过直接作用CDK1来抑制肿瘤进展[14];CDK1的表达可被嗜酸乳杆菌CICC 6074 S层蛋白下调,阻止G1细胞周期,从而发挥其对结肠癌细胞的细胞毒活性[15];CDK1可作为PKN蛋白磷酸化的对应激酶,促进肿瘤细胞贴壁依耐性生长和迁移,充当原癌基因的作用[16];在骨肉瘤中,Huang等[17]研究证实,CDK1在骨肉瘤组织的细胞质中阳性表达,被确定是miR-199a-3p的潜在靶基因。在甲状腺癌中,研究者使用组织芯片证实CDK1蛋白在甲状腺癌(THCA)组织中的表达明显高于在非肿瘤组织中的表达,CDK1基因在THCA组织中共表达的KEGG分析表明:细胞周期、甲状腺激素合成、引起自身免疫性甲状腺疾病等是CDK1在甲状腺癌中表达最丰富的途径[18]。在乳腺癌中,由于选择性阻断CDK1单独或与其他治疗药物联合使用与有效的抗癌效果有关,因此CDK1可能被认为是乳腺癌治疗的靶点之一[19]。在腺样囊性癌和非小细胞肺癌中的研究表明,CDK1的高表达与癌症患者的总体生存率较低相关,因此CDK1可作为诊断和预后的肿瘤标志物或药物治疗靶点之一[20-21];在宫颈癌中,Luo等[22]的研究揭示了CDK1在宫颈癌发展过程中对基因相互作用网络的综合作用,从而表明CDK1作为治疗靶点的潜在作用。此外,有研究[23]表明,CDK1活跃于多种肿瘤调节细胞粘附的细胞周期,可作为多种癌症的临床预后生物标志物。
在HCC中,CDK1的异常表达可以调节凋亡素诱导的凋亡,在肿瘤进展中起着关键作用[24]。CDK1的过度表达也被发现与HCC的门脉侵犯、甲胎蛋白水平高和预后不良直接相关[25]。最近的一项研究发现二甲双胍可以通过诱导G2/M期阻滞来显著抑制HCC细胞的增殖,并能有效地降低CDK1的表达[26],提示CDK1可能参与了HCC细胞周期中的细胞增殖过程。另一项研究表明miR-582-5p通过直接抑制CDK1和Akt3的表达,间接抑制cyclin D1的表达来调控HCC的进展[27],Wang等[28]综合生物信息学分析发现,CDK1、CCNB1、CCNB2、MAD2L1和TOP2A等5个HUB基因可作为预测肝癌预后的生物标志物;Sun等[29]应用生物信息学分析筛选发现CCNB1、CDK1、RRM2和BUB1B在肝癌组织中的过度表达与肝癌患者的不良生存相关,这些基因可能成为肝癌治疗的潜在靶点;He等[30]通过生物信息学发现CDK1可能通过细胞周期和p53信号通路在肝硬化转化为HCC过程中发挥重要作用。Zou等[31]发现CDK1、CCNB1和CCNB2是HCC潜在的预后生物标志物,并与HCC免疫细胞浸润有关。
在HCC中,通过3组芯片共369例HCC组 织和160例癌旁组织的基因表达量分析发现,CDK1在HCC组织中的表达明显高于癌旁组织,后期的生存分析曲线显示CDK1高表达患者的生存时间较CDK1低表达患者明显减少,但上述实验数据均来源于生物信息学,由于其固有缺陷如平台与样本选择的不同可产生一定的差异,且数据信息混杂、缺乏足够实验基础,所以最后在本研究中心选取70例HCC组织和癌旁组织进行表达验证,结果为CDK1在HCC组织中的评分为(7.871 8±1.524 87)分,在癌旁组织中的评分为(3.410 3±1.163 43)分,两组进行比较,差异具有统计学意义(t=14.429,P<0.0001),与预期结果相符。
综上所述,本研究基于生物信息学分析发现CDK1、ASPM、RRM2、TOP2A、CENPF、CCNB1、PTTG1、ECT2、CDKN3基因可能是HCC发生、发展的重要基因,且涉及细胞有丝分裂的负调控、细胞周期、核染色体隔离和雌配子的产生等方面,最后,选用CDK1在HCC组织和癌旁组织中进行验证,发现在HCC组织中CDK1的表达高于癌旁组织,与本研究预期结果相符。生物信息学在发现新靶点方面具有强大功能,但是需要分子生物学、细胞生物学、临床实验及疗效方面的研究来验证。这9个HCC相关基因均在HCC的发生、发展过程中具有巨大的作用,有望成为HCC筛查及治疗的新靶点,同时也将为研究HCC的发生、发展提供一定的理论基础。
