两样本孟德尔随机化方法分析血液代谢物与冠心病的因果关系
2021-02-27王子贤赖伟华钟诗龙
王子贤,赖伟华,钟诗龙
1华南理工大学生物科学与工程学院,广东 广州510006;2广东省人民医院//广东省医学科学院药学部,广东广州510080
冠状动脉粥样硬化性心脏病,简称冠心病,是世界范围内最常见的心血管疾病,也是导致患者死亡的主要原因之一[1]。虽然药物和手术治疗在近年来取得了巨大的进步,但是冠心病患者的死亡率仍然很高[2-4]。因此,了解冠心病的发病机制和识别新的预防靶点对冠心病的防治具有重要意义。
血液中的代谢物作为一种环境暴露下的功能中间体,其往往可以反应个体的遗传组成并能预测或影响疾病的发生发展[5]。近年来,血液代谢组学的研究已为预测心血管疾病的发生提供了众多的生物标志物并建立了可靠的预测模型[6-8]。但是,许多代谢物往往只能提供与疾病发生的关联性,其因果关系尚不明确。孟德尔随机化(MR)作为一种流行的遗传流行病学研究设计方法,它通过使用遗传变异作为工具变量,可以探究暴露和结局之间的因果关系[9]。然而到目前为止,在探究与冠心病发生风险相关的MR分析中,其关注的暴露因素大多是广泛表型,如身高[10]、腰围[11]、臀围[12]等,鲜有关注血液代谢物这样的暴露因素集合。基于代谢组学的全基因组关联研究(mGWAS)是一种识别代谢物数量性状位点[13]以了解疾病相关遗传变异的代谢背景的有效途径。Shin等于2014年发表了迄今为止最大规模的mGWAS,绘制了人类血液代谢物的遗传图谱[14],为血液代谢组学的遗传基础提供重要参考价值。Yang等[15]已经利用两样本MR的分析方法评估了这些血液中的代谢物与5种主要的精神疾病之间的因果关系,并成功地发现了两个代谢物与精神分裂症和注意缺陷或多动障碍之间存在稳健的因果关系,为该类疾病的预测和治疗提供了重要的参考。基于此方法来探究这些血液代谢物与冠心病发生风险之间的因果关系。有助于更深入地了解冠心病的发病机制,并可能为冠心病患者的临床诊疗提供新的见解,但目前尚未有相关的报道。
因此,本研究从分子机制角度出发,采用两样本MR分析方法,使用上述大规模的mGWAS数据为暴露文件,以及另一项超大规模的冠心病GWAS数据为结局文件,以探究这些血液中的代谢物与发生冠心病之间的因果关系。本研究具有一定的理论依据和临床转化价值,研究结果可以为指导冠心病的风险预测和治疗工具的开发提供参考。
1 资料和方法
1.1 资料
本研究采用2014年Shin等[14]发表于Nature Genetics上的迄今为止最大规模的mGWAS数据作为暴露文件。该数据是一项包含7824例欧洲人的荟萃分析数据,经过严格的质量控制,共210万个SNP位点和486种血液代谢物(其中包含309种已知代谢物和177种未知代谢物)用于全基因组关联分析。这些代谢物可分为8种代谢物大类:碳水化合物、氨基酸、核苷酸、辅因子和维生素、脂类、肽类、能量产物和异源性生物代谢产物。所有关联分析的汇总数据可在数据库网站公开获取:http://metabo1omics.he1mho1tz-muenchen.de/gwas/.
冠心病的GWAS 数据来源于2011 年Schunkert等[16]发表于Nature Genetics 的一项包含22个独立研究的超大规模的冠心病荟萃分析数据,样本来自欧洲人群的22 233例冠心病患者和64 762例健康人,共有约240万个SNP 位点用于关联分析。数据收录在CARDIoGRAMp1usC4D,可在数据库网站公开获取:http://www.cardiogramp1usc4d.org/.
1.2 工具变量的选择
我们对486种代谢物的遗传变异采用了统一的入选标准。选择MR分析中常用的较为宽松的阈值,即P<1×10-5为显著的关联分析结果入选条件;在提取出每个代谢物对应的显著的SNP后,以千人基因组中欧洲人(EUR)基因型为参考模板,进行连锁不平衡分析,同时满足以下三个条件认为连锁不平衡并保留P值最小的SNP作为独立的遗传变异:(1)位于同个染色体;(2)相互距离在500 kb以内;(3)连锁不平衡参数r2>0.1。
因为处于同个代谢通路的代谢物可能会受到相似的遗传变异调控,即可能存在多个代谢物与同一个SNP显著相关,这将违反MR假设标准。因此,本研究采用限制性选取工具变量的方法[17],排除与两个以上代谢物均显著相关的SNP。同时排除已知的冠心病风险因素相关的SNP(包括身体质量指数[18]、身高[10]、腰围[11]、臀围[12]、腰臀比[19]、血脂[20]相关的SNP)。
1.3 统计分析
本研究涉及的暴露因素为血液中的代谢物,数量众多,与既往的单一暴露因素的MR研究相比,有更为巨大的工作量。因此,本研究中通过编写Per1代码、R代码和She11代码进行批量处理,即依次探究每个代谢物与冠心病的因果关系。其中,连锁不平衡分析采用PLINK(version 1.9)软件[21];MR分析、基因多效性检验以及敏感性分析采用R 中的TwoSamp1eMR 软件包(version 0.4.22)[22]于Linux系统上进行分析。
1.3.1 MR分析 本研究采用逆方差加权法(IVW)[23]作为首要的因果效应估计。IVW法是一种较为理想状态下的估计,是假设在所有遗传变异都是有效工具变量的基本前提下进行的有效分析,具有较强的因果关系检测能力。但是IVW法特别要求遗传变异仅通过研究中的暴露影响目标结局。尽管此研究已尽可能排除了已知的混杂的SNP,然而仍然有许多未知混杂因素会导致基因多效性并对效应值的估计产生偏倚。因此,我们采用了另外4种方法来检验结果的可靠性和稳定性,即MREgger 回归[24]、加权中位数法(WME)[25]、基于众数的简单估计[26]、基于众数的加权估计[26]。依次对每个代谢物进行MR分析,如果以上五种不同的MR模型对因果效应产生了相似的估计值,我们则认为该代谢物与冠心病的因果关系是稳定且可靠的。
IVW法分析的结果中,我们采用严格的多重假设检验的阈值P<1.03×10-4(P<0.05/486)来检验显著的因果关系。并且同时关注P值大于等于1.03×10-4但小于0.05值的代谢物来作为冠心病潜在风险预测因子。P值小于0.05的因果关系将进行如下的异质性检验和基因多效性检验。
1.3.2 异质性检验 由于不同分析平台、实验条件、入选人群的以及SNP的差异,两样本MR分析法可能存在异质性,从而对因果效应的估计产生偏倚。因此,本研究中对主要的IVW分析法和MR-Egger回归采取异质性检验,检验的结果中P值大于0.05则认为纳入的工具变量不存在异质性,可以忽略异质性对因果效应估计产生的影响。
1.3.3 基因多效性检验 MR分析的假设之一是工具变量只能通过暴露影响结局,若工具变量不通过影响暴露而直接影响结局则违背了MR思想,所以需要检验暴露与结局之间的因果推断是否存在基因多效性。采用
MR-Egger回归分析可以来评价基因多效性产生的偏倚,其回归截距可以评估多效性的大小,截距越接近于0,则基因多效性的可能性越小。本研究中通过判断基因多效性检验的P值来衡量分析中是否存在基因多效性,若P>0.05,则认为因果分析中基因多效性的可能性较弱,可以忽略其产生的影响。
1.3.4 敏感性分析 除了采用上述4种方法(MR-Egger回归法、加权中位数法、基于众数的简单估计法、基于众数的加权估计法)来检验结果的可靠性和稳定性。本研究还采用1eave-one-out 法来进行敏感性分析。即对IVW法中P值小于0.05,并且通过了异质性检验和基因多效性检验的代谢物,逐一去除各个相关的SNP并计算剩余的SNP的合并效应,以评估各个SNP对于代谢物的影响。
2 结果
2.1 工具变量(SNP)信息
在486个代谢物中,与之关联的P<1×10-5的共有39 142个SNP,连锁不平衡分析后,得到10 905个独立的SNP,其中,有447个SNP至少与两个代谢物显著相关。另外,与冠心病风险因素相关的SNP共有319个,这些SNP均不包含在本研究中。排除混杂的SNP后,共10 458个SNP纳入后续分析,这些SNP在冠心病的GWAS数据中存在9108个,位点覆盖率为87%。工具变量的质控流程图如图1所示。每个代谢物对应的工具变量的数量的中位数为13,后续分析中排除拥有工具变量的数量小于等于3的5个代谢物和大于等于100的5个代谢物。
2.2 MR分析结果
本研究采用IVW法作为首要的评估代谢物与冠心病之间因果关系的方法。共有32个代谢物与冠心病的因果关系效应值达到名义上显著(P<0.05),其中,包含已知代谢物11个,未知代谢物21个;未发现多重假设检验(P<1.03×10-4)后仍然显著的代谢物(表1)。
在11个已知的代谢物中,包括4种可能与增加冠心病发生风险相关的代谢物,即犬尿氨酸,γ-谷氨酰异亮氨酸,丁二酰基肉碱,血红素;7种可能与降低冠心病发生风险相关的代谢物,即1-甲基黄嘌呤,溶血磷脂酰胆碱,(Des-Arg9)-缓激肽,N-乙酰鸟氨酸,肉豆蔻酸酯,甘氨酸,甘露醇。
2.3 结果的可靠性和稳定性评估
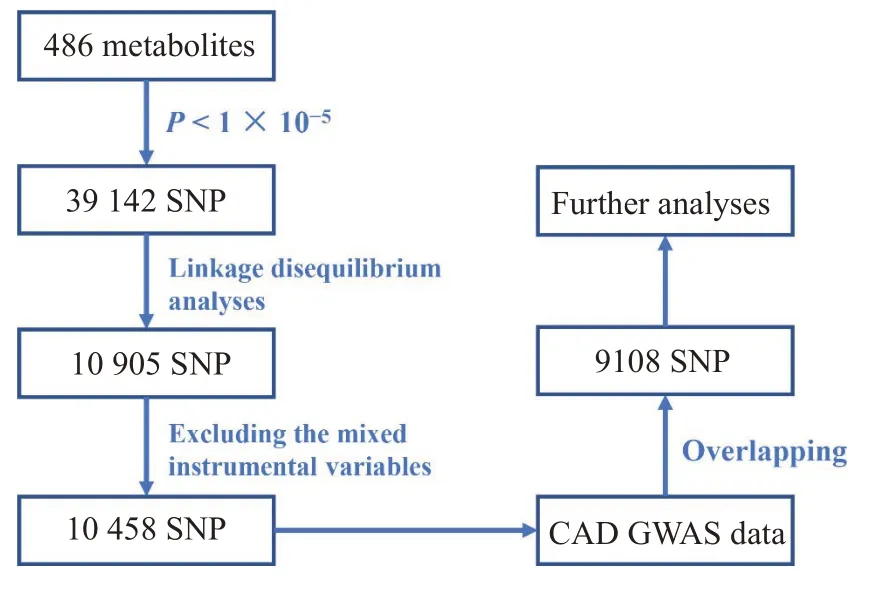
图1 用于MR分析的工具变量质控流程图Fig.1 Flow chart for quality control of the instrumental variables for MR analyses.
对以上11个已知的代谢物所对应的工具变量进行异质性检验和基因多效性检验,排除2个存在异质性或基因多效性的代谢物(犬尿氨酸和1-甲基黄嘌呤)。五种MR模型、异质性检验和基因多效性检验结果见表2。剩余的9个代谢物中,有3个代谢物对应的5种MR模型中有至少3种模型的P值小于0.05[N-乙酰鸟氨酸,(Des-Arg9)-缓激肽,丁二酰基肉碱]。尽管五种MR模型都未能全部达到统计学意义上显著,但是它们均具有相似的效应值,其原因可能是因为IVW法比其他四种MR模型拥有更高的检验效能。3个代谢物的MR分析结果散点图见图2。
用1eave-one-out法对以上3个代谢物[N-乙酰鸟氨酸,(Des-Arg9)-缓激肽,丁二酰基肉碱]的因果效应进行敏感性分析。3个代谢物中都存在至少1个SNP对结果的效应值产生显著影响,因此,对这些SNP进行剔除后,我们重新对这3个代谢物进行了MR分析。N-乙酰鸟氨酸,(Des-Arg9)-缓激肽和二酰基肉碱的IVW法对应的效应值均不再显著(表3)。
3 讨论
本研究运用了公共数据库中大规模的mGWAS和GWAS数据,采用无偏倚的两样本孟德尔随机化分析方法探究了486种血液代谢物与冠心病发生风险之间的因果关系。然而经过严格的质量控制,尚未找到非常有力的证据表明这些血液代谢物与冠心病的发生之间存在直接的因果关联。
此研究中涉及的486种血液中的代谢物,尽管均未能通过多重假设检验的阈值,但仍然为我们提供了11种潜在的冠心病风险预测因子。包括4种可能与增加冠心病发生风险相关的代谢物(犬尿氨酸,γ-谷氨酰异亮氨酸,丁二酰基肉碱,血红素)。其中包含的3个代谢物[即,N-乙酰鸟氨酸,(Des-Arg9)-缓激肽,丁二酰基肉碱]在至少3种孟德尔随机化模型中都达到了统计学显著。本研究发现N-乙酰鸟氨酸和(Des-Arg9)-缓激肽可能作为潜在的保护性物质从而降低冠心病的发生风险。N-乙酰鸟氨酸是人血去蛋白血浆中的次要成分,暂未发现其关于心血管研究的报道。缓激肽作为一种血管活性激肽,具有改善心功能并可以降低远期心脏事件的作用[27]。实验研究表明,缺乏缓激肽B2受体基因的小鼠更容易出现高血压,心脏肥大和心肌损伤[28]。由此可见,(Des-Arg9)-缓激肽在保护心血管疾病和降低冠心病发生风险之间可能具有重要作用。丁二酰基肉碱是来自血液和肝脏中能量代谢的中间体,已有文献报道其是心血管疾病的危险因素之一[29],然而其与冠心病发生之间的因果关系仍然需要后续的进一步研究来证实。
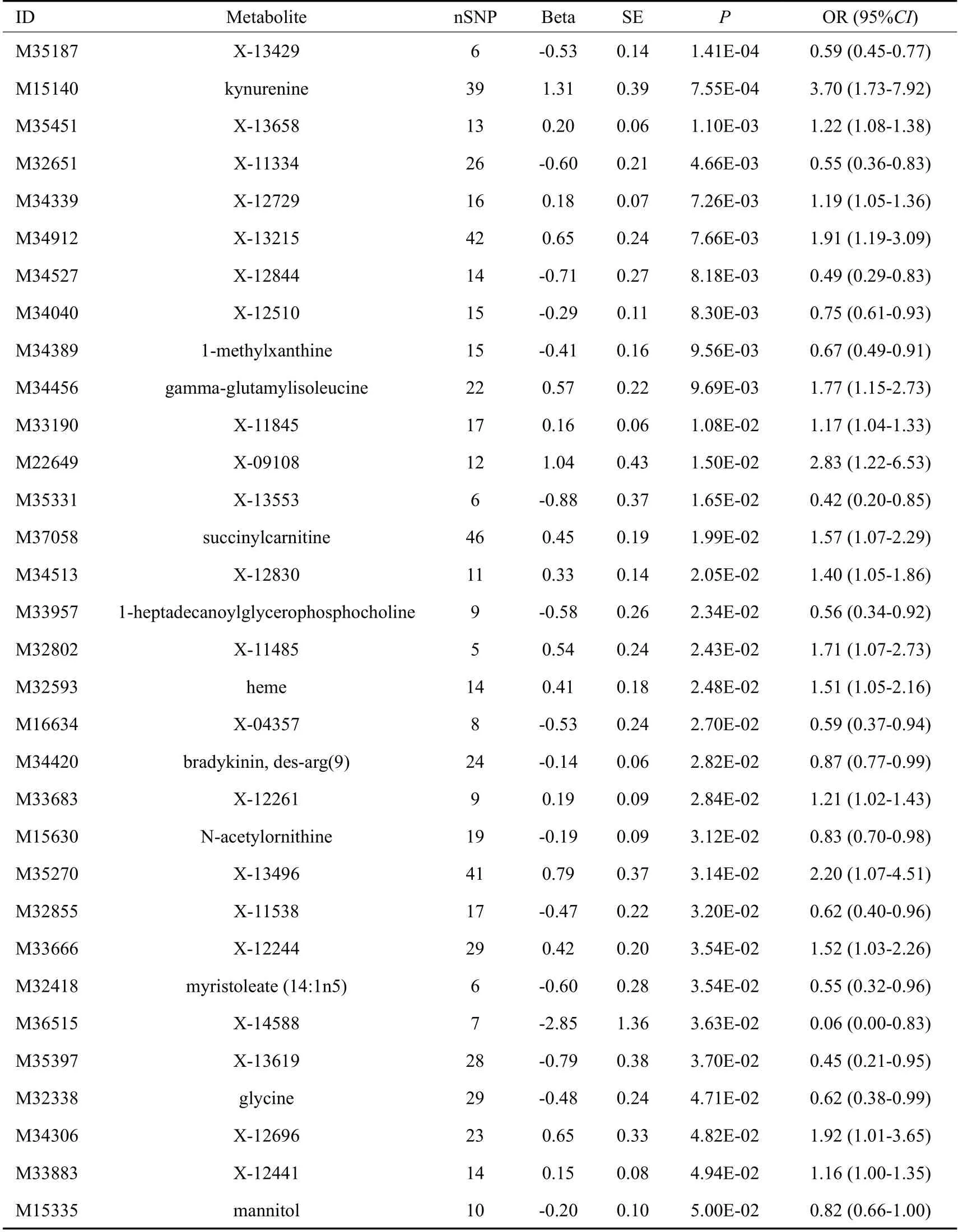
表1 IVW方法达名义上显著的代谢物对应的效应值结果Tab.1 Nominally significant results of IVW method
本研究具有如下创新性:(1)本研究从分子机制角度出发,以血液中的代谢物为暴露因素,探究其与冠心病发生风险之间的因果关系,具有较强理论依据和重要临床研究价值;(2)本研究采用严格的质控条件和分析方法,运用多种模型来评估因果效应,研究结果具有可靠性和稳定性;(3)与既往的单一暴露因素的孟德尔随机化研究相比,本研究中涉及的暴露因素为血液中的代谢物,数量众多,具有十分巨大的工作量和分析挑战性。本研究中也存在一定的局限性:(1)mGWAS数据和冠心病的GWAS数据均来源于欧洲人群,后续更为全面的研究仍需在不同人种之间展开;(2)初步分析得到的冠心病风险预测因子大多为未知代谢物,其功能结构存在不确定性;(3)尽管我们采用迄今为止最大规模的mGWAS数据,后续研究仍然需要进一步扩大的样本量来为代谢物的遗传影响提供更准确的评估。
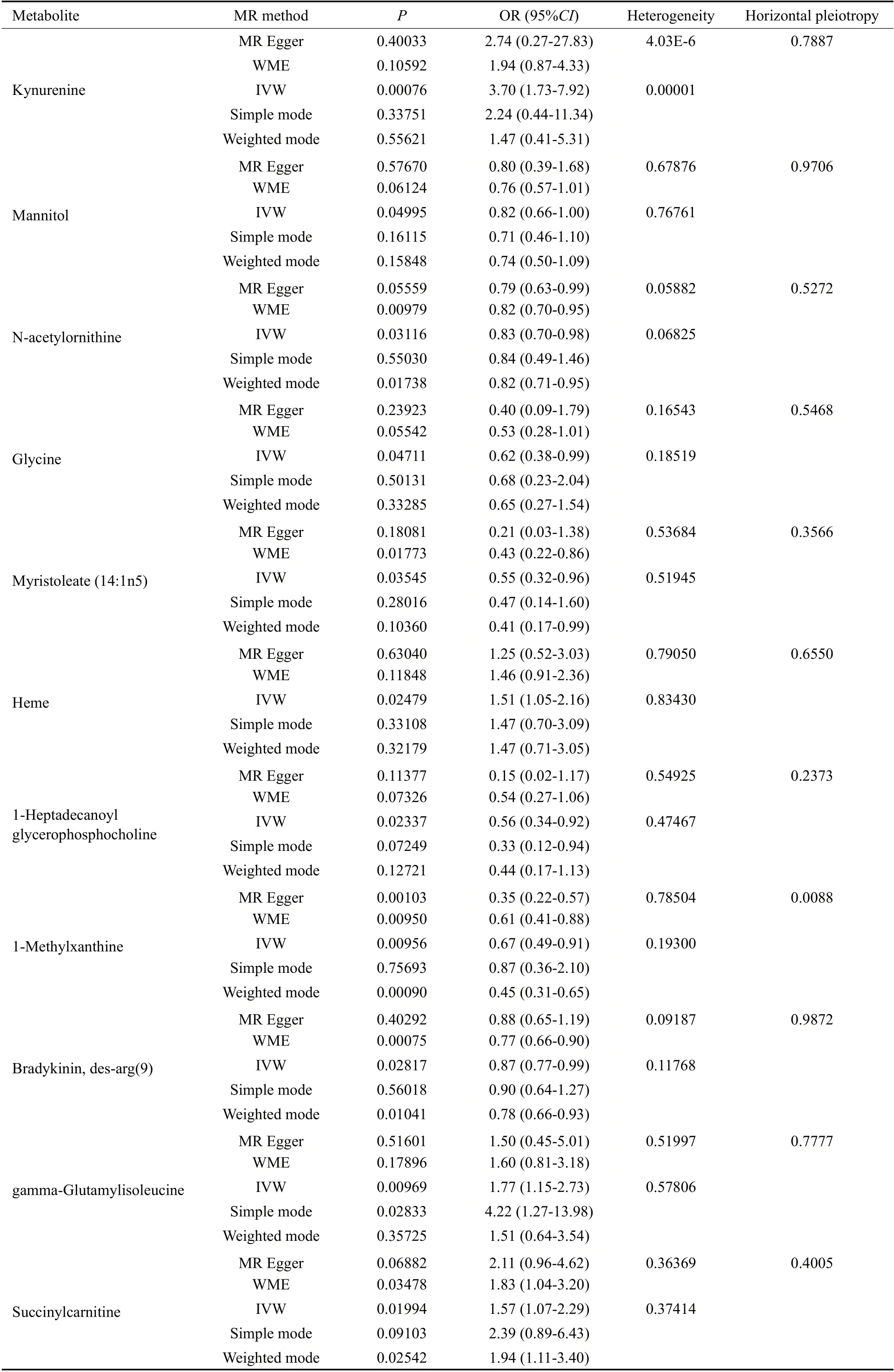
表2 已知代谢物的5种MR分析以及异质性检验和基因多效性检验结果Tab.2 Results of 5 MR models of known metabolites and the heterogeneity and pleiotropy tests
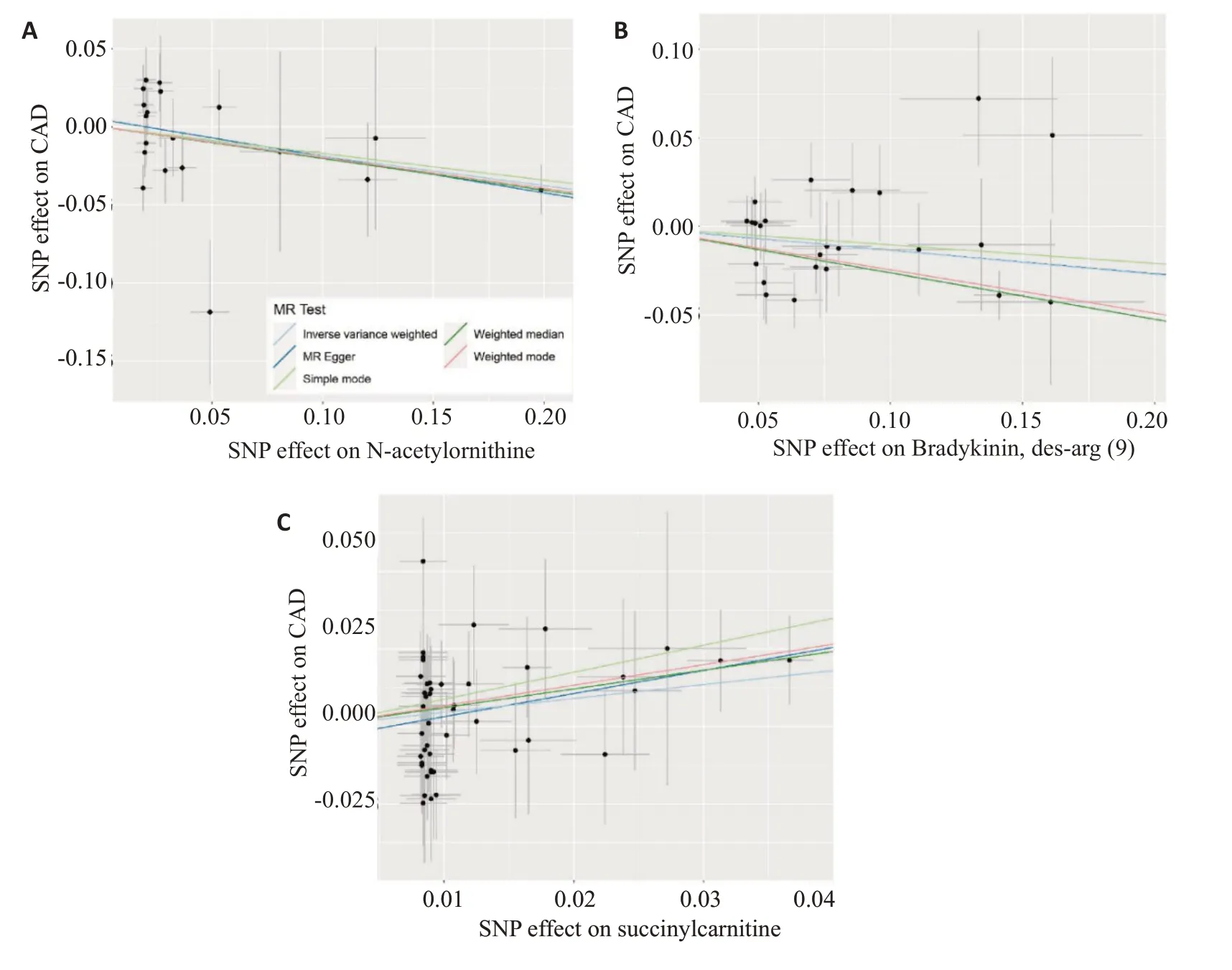
图2 三个具有潜在因果关系的代谢物的5种MR模型散点图Fig.2 Scatter plots of the 5 MR models for 3 metabolites with potential causal relationship with CAD.A:Nacetylornithine;B:Bradykinin,des-arg(9);C:Succinylcarnitine.

表3 Leave-one-out法检验剔除混杂SNP后的3种代谢物MR分析结果Tab.3 MR analysis results of the 3 metabolites after removing mixed SNP by leave-one-out method
综上,我们采用了两样本孟德尔随机化的方法探究了486种血液代谢物与冠心病的因果关系。尽管没有发现这些血液代谢物与冠心病发生风险之间存在稳健的因果关系,但本研究中发现的潜在的冠心病风险预测因子仍为揭示遗传-暴露相互作用在冠心病发病机制中的作用提供了新的见解。
