外电场对水在CaO(100)表面吸附的第一性原理计算
2021-02-26闪静祎王军凯黄珍霞戴新宇胡前库周爱国
闪静祎 王军凯 黄珍霞 戴新宇 胡前库 周爱国
1)河南理工大学 材料科学与工程学院 河南焦作454003
2)河南理工大学 化学化工学院 河南焦作454003
镁钙系耐火材料具有耐高温、抗渣性好、抗热震性优良等优点,广泛应用于连铸中间包、炉外精炼钢包、电炉、转炉内衬及大型干法水泥窑烧成带等[1-4]。但是镁钙系耐火材料也存在巨大的缺点,即其中的游离氧化钙极易水化,导致耐火材料损毁。因此,如何抑制镁钙系耐火材料中游离氧化钙的水化是一个关键的问题。
目前,国内外研究抑制镁钙系耐火材料水化的方法主要包括煅烧法[5-6]、表面处理法[7-8]、添加剂法[9-11]、密封包装法和生产工艺控制法。但高温煅烧法能耗高,对煅烧设备以及环境温度要求较高,工业化生产有一定难度[12],而使用二步煅烧法会使经过煅烧循环后的氧化钙颗粒内部孔结构变得更加开放,导致极易与外界物质发生反应[13]。表面处理法在使用过程中氧化钙表面形成的薄膜易分解,不能从根本上解决镁钙系耐火材料中游离氧化钙易水化的问题[14]。添加剂法在引入添加剂的同时会引入一些低熔点物质降低镁钙系耐火材料的使用性能[15-16]。密封包装法和生产工艺控制法只适用防止镁钙系耐火材料在运输过程中的水化,且包装易破损,这也不能从根本上解决水化问题[17]。因此,抑制镁钙耐火材料水化必须从氧化钙水化机制进行新的探索。
近年来,随着计算化学的发展和应用,密度泛函理论(DFT)被广泛应用于化学、物理、材料和生物等领域。通过计算不仅可以获得材料的稳定结构,还可以揭示反应的深层机制,深入理解反应过程。文献[18-20]表明,密度泛函理论计算已经应用于水分子在CaO表面吸附问题的研究。Wei等[18]采用密度泛函理论计算研究了H2O分子在CaO(100)面上的吸附。结果表明:H2O与CaO表面的相互作用很强,属于化学吸附。Han等[19]采用密度泛函理论计算也得到了类似的结果。Ma等[20]同样采用密度泛函理论计算研究CaO与H2O分子的相互作用,结果发现在CaO表面吸附H2O分子后,CaO中的O原子与H2O中的H原子之间形成共价键。这些研究表明,CaO的水化过程与H2O分子在CaO表面的吸附行为密切相关。解决镁钙系耐火材料的水化问题,就必须抑制H2O分子在CaO表面的吸附。
研究表明,外加电场是控制气体小分子在固体表面吸附的一种有效手段。Liang等[21]在Ga掺杂石墨烯表面施加正电场,使NO2分子在其表面的吸附能增加35%,说明外加电场可以抑制NO2气体分子在Ga掺杂石墨烯表面的吸附,从而对NO2小分子起到脱附作用。Tu等[22]在外加电场条件下,研究了水分子在Cd(0001)表面的吸附行为。结果表明,与不加电场相比,正电场存在的条件下,H2O分子在Cd(0001)表面的吸附会增强,负电场下则会减弱。Wang等[23]在g-C3N4(001)表面添加电场,并通过控制外加电场强度实现了选择性捕获和分离H2S、H2O及CO2分子。同样许多试验和理论研究也表明[24-26],施加外部电场会大大影响固体材料表面的吸附能力。
目前,关于外加电场条件下水分子在氧化钙表面的吸附行为尚缺乏相关的研究。基于此,在本工作中首先研究了无外加电场条件下,氧化钙表面结构和水分子构型对H2O分子在CaO(100)表面吸附行为的影响,确立了H2O分子在CaO(100)表面最佳吸附构型。在此基础上,又分别研究了外加正、负电场条件下,水分子在CaO(100)表面的吸附行为。
1 计算方法
所有的第一性原理量子化学计算都是在Materials Studio软件包中的DMol3模块[27]下进行的。利用Perdew-Burke-Ernzerhof(PBE)泛函的广义梯度近似(GGA)进行几何优化[28-29],研究了H2O分子在CaO(100)表面的吸附。所有计算采用双数值加速极化(DNP)基组,其结果可与Pople的6-31G(d,p)基组相媲美。采用Grimme的DFT-D用于分子间相互作用的长距离色散校正[30]。在几何优化过程中,能量收敛标准是2.721 14×10-4eV,最大力收敛标准是0.544 228 eV·nm-1,最大位移收敛标准是5×10-4nm。采用Monkhorst-Pack方法[31]对布里渊区进行取样,几何优化过程采用5×5×1的K点网格。此外,因为CaO(100)面的表面能最低,且容易暴露在外[32],所以,在本工作中研究CaO(100)面。建立CaO(100)面表面模型,固定下面三层原子,只允许上面三层原子自由移动。为了避免相邻表面结构间的相互作用,使用3 nm的真空层高度(示意图见图1)。
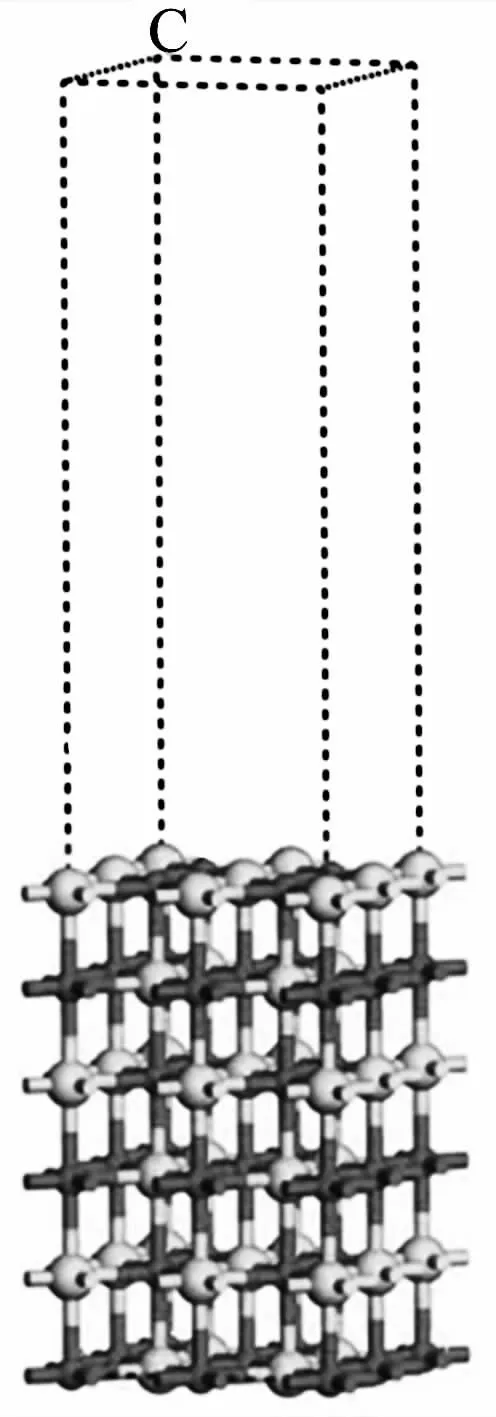
图1 CaO(100)表面示意图Fig.1 Sketch of CaO(100)face
从吸附能的角度分析H2O分子在CaO(100)表面上的吸附稳定性:
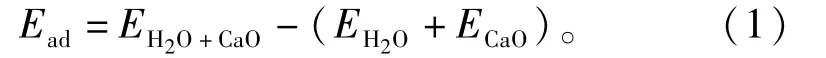
其中,EH2O+CaO、EH2O、ECaO分别为H2O 分子与CaO(100)表面吸附后体系的总能量、CaO(100)表面结构的能量、孤立的H2O分子的能量。吸附能越负,表明吸附体系越稳定。此外,采用Mulliken电荷分布研究了H2O分子与CaO(100)表面间的电荷转移[33]。
2 结果与讨论
2.1 水分子在CaO(100)表面的吸附
2.1.1 氧化钙表面结构对水分子吸附的影响
首先研究了氧化钙表面结构对水分子吸附行为的影响。考虑了CaO(100)表面的3种吸附位点,分别是:由钙原子组成的正方形中心上方的空心位点(H)、钙-钙键中心上方的桥位点(B)和钙原子正上方的顶部位点(T)。H2O分子在CaO(100)表面的3种初始吸附构型和经过充分优化后的最终吸附模型示意图如图2所示,H2O分子与CaO(100)表面之间的初始距离设为0.300 0 nm。对比优化前后的模型图可以发现:1)吸附在H、B和T位点的H2O分子与CaO(100)表面间的距离缩短,由最初的0.300 0 nm分别变为0.199 4、0.185 0和0.255 3 nm,表明CaO(100)表面对不同吸附位点的H2O分子均表现出一定的吸附作用。2)吸附在B和T位点的H2O分子经过几何优化后仍吸附在原位置。而初始吸附在H位点的H2O分子经过优化后则移向B位点。这表明B位点可能比H位点更容易吸附H2O分子。3)3种吸附位点中,B位点的H2O 分子优化后距离CaO(100)表面最近,而T位点的H2O分子最远,再次表明B位点可能最容易吸附H2O分子。
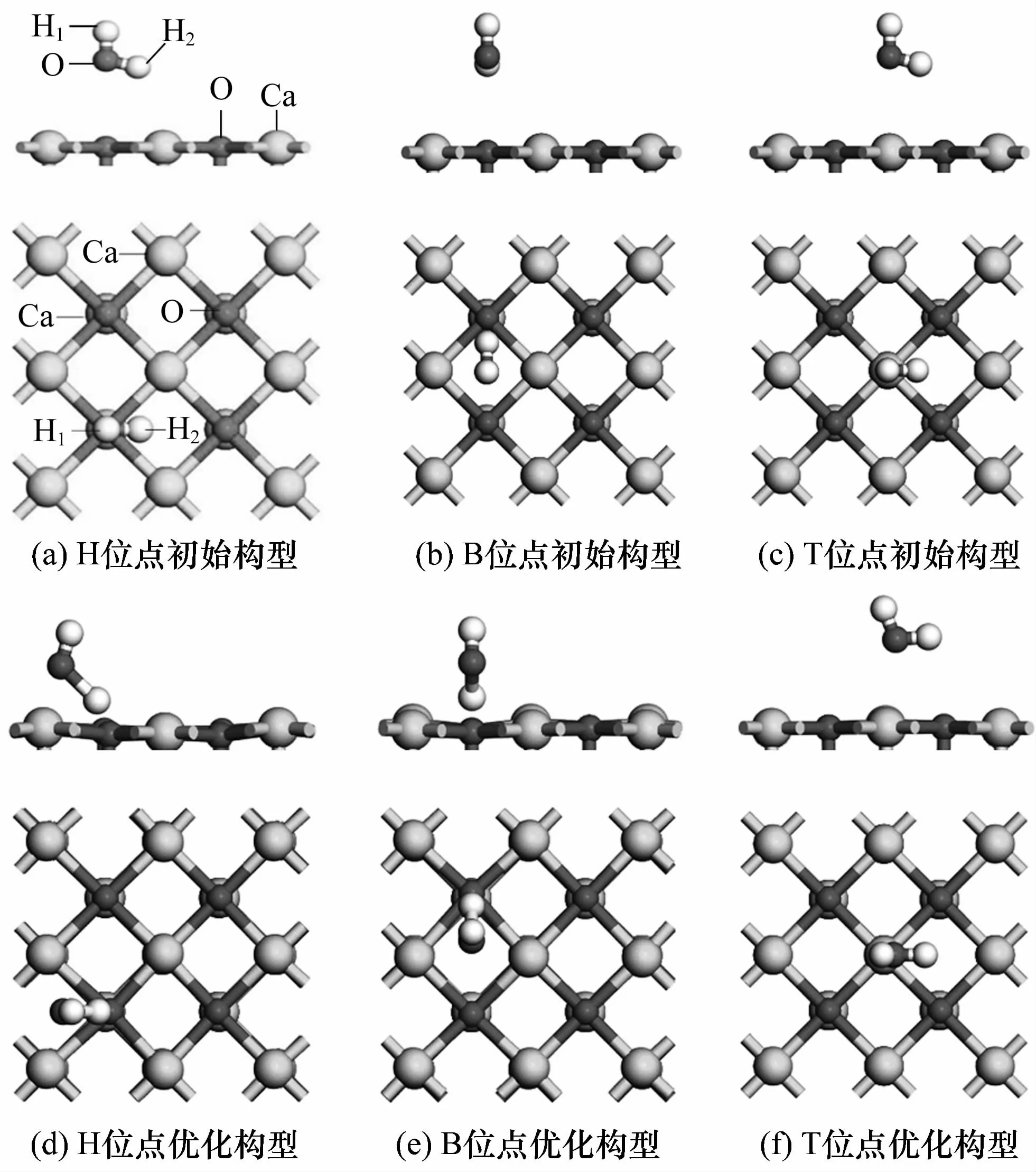
图2 H2O分子吸附在CaO(100)表面H、B、T位点的初始构型和优化构型示意图Fig.2 Initial and optimized configurations of hollow(H),bridge(B)and top(T)sites adsorbed by H2O on CaO(100)face
为进一步研究氧化钙表面结构对水分子吸附行为的影响,又计算了不同吸附位点下H2O分子与CaO(100)面间的吸附能和电荷转移情况,结果如表1所示。

表1 不同吸附位点吸附能和电荷的值Table 1 Value of adsorption energy and charge at different sites
从中可以看出:1)吸附在H、B和T位点的H2O分子与CaO(100)表面间的吸附能均为负值,表明CaO(100)表面的不同位点均可以吸附H2O分子。2)H2O分子吸附在CaO(100)表面3种位点时的吸附能从小到大依次为B<H<T,说明H2O分子在B位点的吸附强于H和T位点,且此时的吸附属于化学吸附。3)H2O分子所带负电荷数量从大到小依次为B=H>T,该结果表明CaO(100)表面的B位点(H2O分子在H位点的吸附会转向B位点)可以向H2O分子转移更多的负电荷,进一步说明H2O分子在B位点的吸附强于T位点。综合以上分析结果可知,CaO(100)表面的B位点最容易吸附H2O分子。该结果与文献[18]的报道一致。
2.1.2 水分子构型对吸附行为的影响
进一步研究了水分子构型对吸附行为的影响。由于H2O分子(H—O—H键角104.5°,H—O键长0.096 9 nm)的三个潜在结合基团(H、O和O—H)均可以接近CaO(100)表面。因此,考虑了5种不同的H2O分子吸附取向,示意图如图3所示。
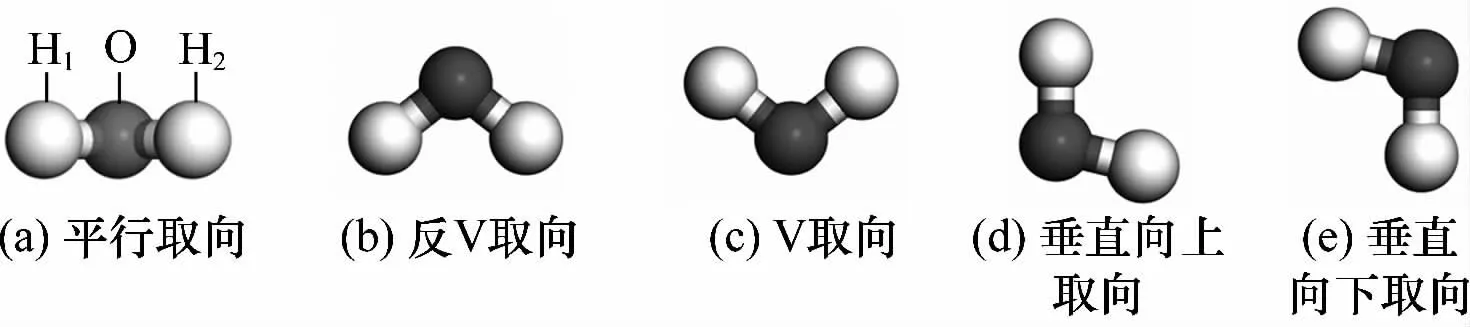
图3 H2O分子不同吸附结构示意图Fig.3 Different adsorption orientations of H2O
由2.1.1部分的研究结果可知,CaO(100)表面的B位点是H2O分子最可能的吸附位置,因此,在CaO(100)表面的B位点建立了上述5种不同取向的H2O分子吸附构型,其结构优化前后的模型示意图如图4所示。从图中可以看出:1)优化后,不同取向的H2O分子与CaO(100)表面的距离发生了明显变化,其中,平行取向、垂直向上取向和垂直向下取向的H2O分子到CaO(100)表面的距离缩短,分别为0.292 2、0.185 0、0.185 4 nm;而反V取向和V取向的H2O 分子到CaO(100)表面的距离则增长为0.383 2、0.302 0 nm。说明与反V取向和V取向相比,平行取向、垂直向上取向和垂直向下取向的水分子可能更容易在CaO(100)表面的B位点吸附;其中,垂直向上取向和垂直向下取向可能最容易被CaO(100)表面B位点吸附。2)由于H2O与CaO(100)面之间的相互作用,垂直向上取向的初始构型和垂直向下取向的初始构型经过优化后得到的吸附模型基本差异不大,结合优化后构型中水分子到CaO(100)表面的距离结果可以推测,这两种构型可能最容易被CaO(100)表面B位点吸附。
不同取向H2O分子在B位点吸附时对应的吸附能、电荷转移、键长变化如表2所示。结果表明:1)不同取向的H2O分子吸附在CaO(100)表面B位点的吸附能均为负值,其中垂直向上取向和垂直向下取向初始构型的吸附能较大,H2O分子容易被吸附。2)优化后,垂直向上取向、垂直向下取向和平行取向的H2O分子获得电荷带负电,其数量由大到小依次为垂直向上取向的=垂直向下取向的>平行取向的;而反V取向和V取向构型的H2O分子则失去电荷带正电,其带电量分别为0.008和0.091 e。可以看出:垂直向上取向和垂直向下取向构型的H2O分子从CaO(100)表面B位点获得的电荷数量相同,进一步表明垂直向上取向和垂直向下取向构型的吸附强于其他构型。3)对初始键长(0.096 9 nm)观察发现,吸附在CaO(100)表面B位点的不同取向H2O分子优化后,平行取向构型、反V取向构型和V取向构型的H1—O键键长和O—H2键键长仍保持相等长度,且与初始键长相比仅有略微增大;而垂直向上取向和垂直向下取向构型中,虽然H2O分子的H1—O键键长变化不大,但是O—H2键键长却显著增大,分别变为0.144 4和0.144 5 nm。该结果表明垂直向上取向和垂直向下取向构型的H2O分子和CaO(100)之间的相互作用更强,进一步表明垂直向上取向和垂直向下取向构型的H2O分子最容易被CaO(100)表面B位点吸附。

图4 吸附在CaO(100)表面B位点的H2O分子的初始和优化构型示意图Fig.4 Initial and optimized configurations of H2O adsorbed on B point of CaO(100)face
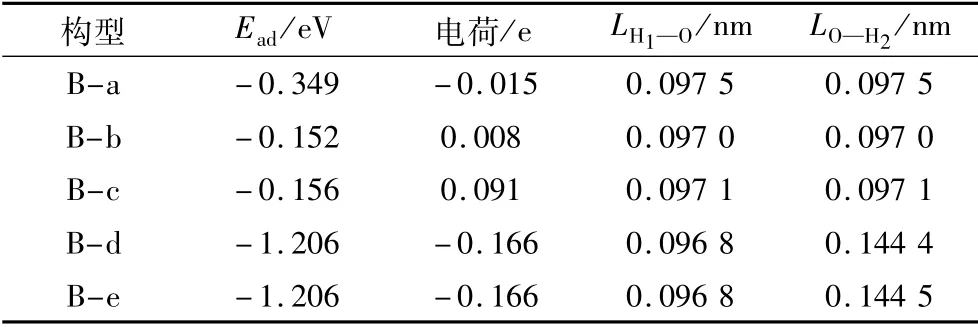
表2 不同取向H2O分子吸附前后的电子电荷、键长、吸附能的值(“-”表示获得电子)Table 2 Values of electron charge,bond length,adsoption energy before and after adsorption of H2O with different orientations(the“-”denotes gaining electrons)
2.2 外加电场对水分子在CaO(100)表面吸附的影响
以上研究表明,B-d和B-e构型是最稳定的吸附结构,且二者尽管初始模型有差异,但优化后的模型基本相同。因此,在本工作中选择其中的B-d构型进一步研究外加电场对水分子吸附的影响。添加垂直于CaO(100)表面的外部电场,规定z轴正方向为正电场,反方向为负电场(示意图见图5)。在-5.142 3 V·nm-1至+5.142 3 V·nm-1的电场下,分别对单独水分子、CaO(100)表面以及水分子吸附在CaO(100)表面上的几何构型进行了优化。
2.2.1 外加负电场
首先研究了负电场对H2O在CaO(100)表面吸附的影响。在不同大小的外加负电场作用下,吸附能、H2O分子键角和二者距离的变化如图6所示。从图6(a)中可以看出,吸附能随着负电场强度的增加而逐渐增大。这表明H2O分子与CaO(100)表面之间的吸附作用随着负电场强度的增加逐渐减弱。从图6(a)中还可以看出,随着负电场强度的增加,H2O分子的键角逐渐减小,趋向于单独水分子键角(104.5°)大小。该结果也表明:随着负电场强度的增加,水分子受CaO(100)表面的影响逐渐减弱。此外,从图6(b)中二者距离的变化曲线可以看出,随着负电场的电场强度增大,H2O分子到CaO(100)表面距离越来越大,当电场强度从0增加到-5.142 3 V·nm-1时,距离从0.191 3 nm逐渐增加到0.207 2 nm(见表3),说明H2O分子逐渐远离CaO(100)表面。
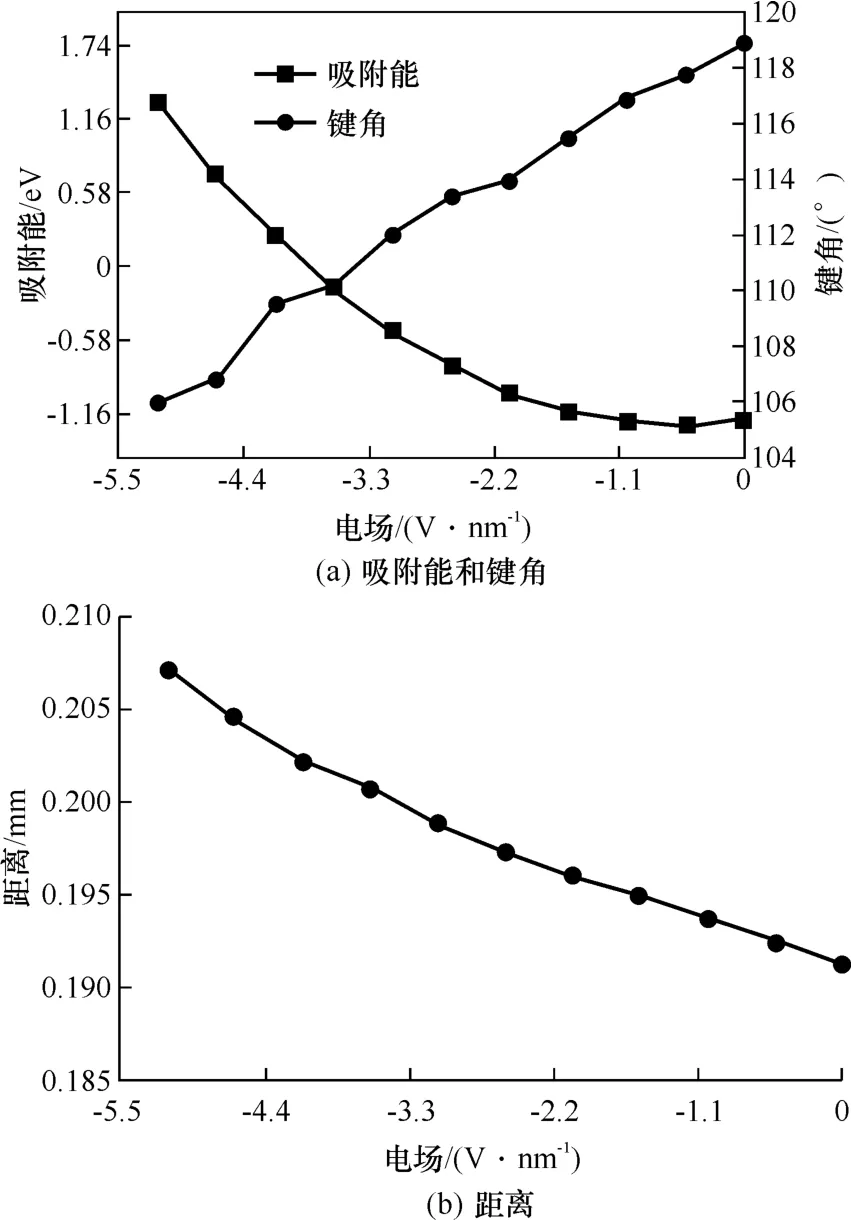
图6 不同负电场作用下的吸附能、键角及距离的变化Fig.6 Variation of adsorption energy,bond angle and distance under different negative electric fields
图7示出了负电场强度E为0、-2.056 9、-3.599 6、-4.113 8及-5.142 3 V·nm-1时对应的吸附构型。结合不同电场下吸附能和二者距离的数值(见表3)可以看出,当电场强度从0增加到-2.056 9 V·nm-1时,吸附能从-1.205 eV变化至-0.979 eV,水分子键角从118.830°减小到113.959°,H2O分子与CaO(100)表面距离从0.191 3 nm增加到0.196 1 nm。表明微弱的负电场虽然可以抑制H2O分子在CaO(100)表面的吸附,但此时的吸附仍然为化学吸附。当负电场强度从-2.056 9 V·nm-1变化到-3.599 6 V·nm-1时,H2O分子进一步远离CaO(100)表面,吸附能继续增加,键角持续减小。这表明此时仅有微弱的吸附作用。当负电场强度从-3.599 6 V·nm-1变化到-5.142 3 V·nm-1时,吸附能由负值变为正值,H2O分子与CaO(100)表面的距离从0.200 8 nm进一步增大至0.207 2 nm,键角从110.210°进一步减小至105.991°,几乎与自由水分子的键角相同。这表明此时H2O分子与CaO(100)表面几乎没有相互作用。
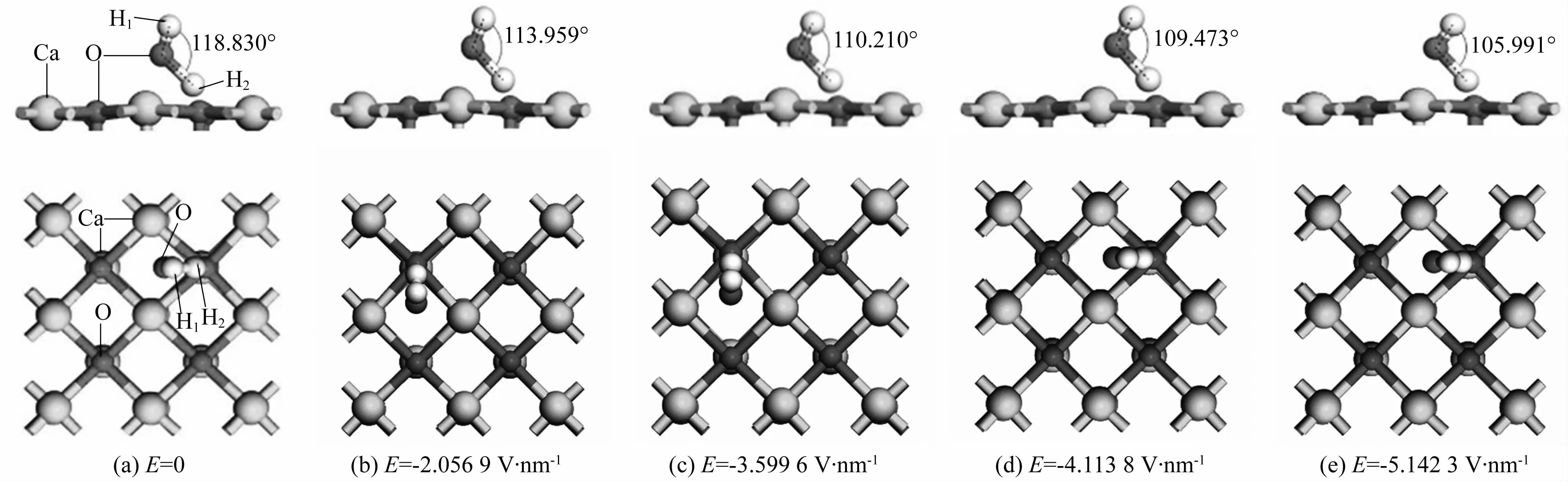
图7 不同电场下,H2O分子吸附在氧化钙上的优化构型示意图Fig.7 Optimized structures of H2O adsorbed on CaO under different electric fields
Ca原子和H2O分子中O原子的电荷变化曲线如图8所示。从图8中可以观察到,随着负电场强度的逐渐增强,Ca原子的电荷逐渐减小,H2O分子中的O原子的电荷也不断减小,说明Ca原子向O原子转移的电子越来越少,即H2O分子与CaO(100)的相互作用不断减弱。以上结果表明,外加负电场对H2O分子在CaO(100)表面的吸附有抑制作用。
2.2.2 外加正电场
研究了外加正电场对H2O分子在CaO(100)面上吸附的影响。不同电场强度作用下H2O分子到CaO(100)表面的距离和二者之间吸附能的变化如图9所示。从距离随电场强度的变化曲线可以看出,随着正电场电场强度的增大,距离基本不变。观察Ead变化曲线可知,在外部正电场下,吸附能会随着电场强度的增加而增大,直至增为正值。具体而言,在无电场时(外加电场为0),H2O分子在CaO(100)表面的吸附能为-1.205 eV,外加正电场为+5.142 3 V·nm-1时,Ead为1.777 eV,比无电场时增加了约247%。先前的研究表明,当吸附能小于-0.50 eV时,气体小分子可以被固体表面有效捕获[35],那么吸附能值越正则表明越不利于小分子在固体材料表面吸附。因此,在外部正电场的作用下,水分子和CaO(100)表面之间的吸附也会变弱。
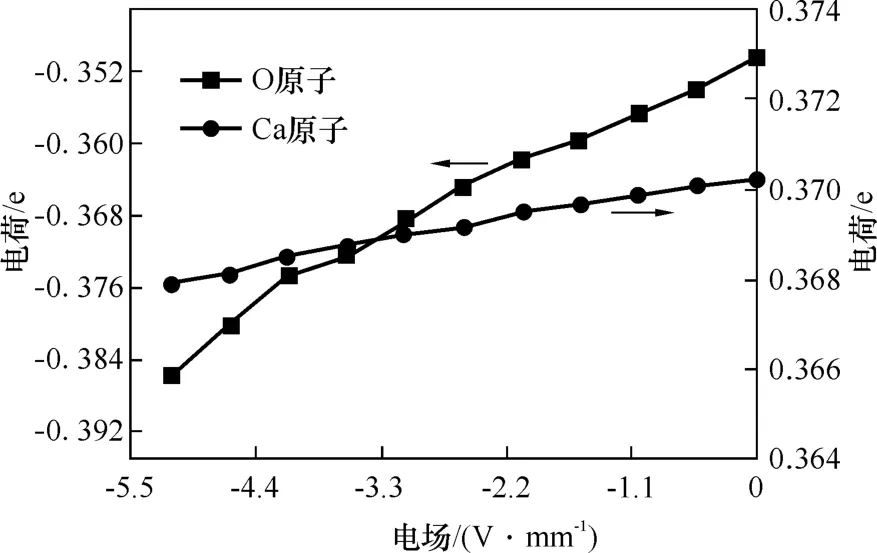
图8 不同电场下O原子和Ca原子的电荷变化Fig.8 Charge changes of O and Ca atoms at different electric fields
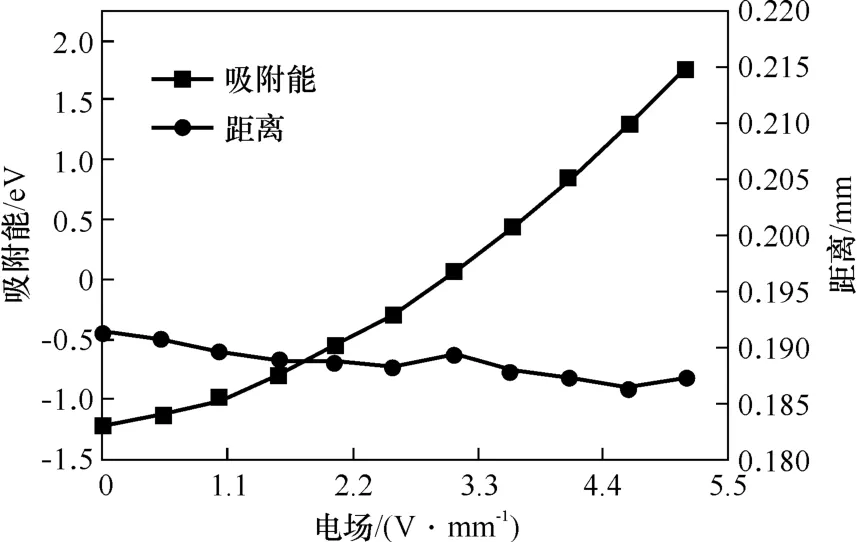
图9 不同正电场下吸附能和距离变化Fig.9 Variation of adsorption energy and distance under different positive electric fields
3 结论
(1)采用DFT计算研究了H2O分子在CaO(100)表面的吸附行为。H2O分子以垂直向上取向构型和垂直向下取向构型吸附于CaO(100)面的B位点时吸附能最负,吸附最稳定。
(2)在外加负电场的作用下,H2O分子与CaO(100)表面之间的距离和吸附能均随电场强度的增强而增大,分别增至0.207 2 nm和1.285 eV。同时,H2O分子的键角不断减小,减小至105.991°,逐渐趋向于单独水分子。电荷转移结果表明,随着负电场强度的增加,Ca原子向O原子转移的电子减少,吸附作用减弱,水分子逐渐远离氧化钙。在外加正电场的作用下,随着电场强度的增加,距离变化不大,但吸附能逐渐由负值变为正值,吸附作用也减弱。因此,外加电场可以抑制H2O分子在CaO(100)表面的吸附,有望为镁钙系耐火材料防水化提供一种新方法。
