β-1,4-葡聚糖内切酶的定向进化
2021-02-25郭成栓杨桂玲刘晓珊
郭成栓,杨桂玲,刘晓珊
(广东食品药品职业学院生物技术学院,广东 广州 510520)
纤维素酶包括β-葡聚糖外切酶、β-葡聚糖内切酶和纤维二糖酶[1-3]。纤维素酶通常在酸性条件下发挥其催化作用,在碱性条件下酶活较低甚至没有,从而限制了其在碱性环境中的应用。碱性纤维素酶是一种缺少β-葡聚糖外切酶和纤维二糖酶的非全纤维素酶组分的β-1,4-葡聚糖内切酶,不会破坏纤维素的结构,可以在碱性条件下发挥催化作用,被广泛应用于洗涤剂、脱墨等行业[4-6]。洗涤剂中加入碱性纤维素酶,一方面可增强洗涤剂的去污能力,有效去除棉织物上的污垢;另一方面还能对棉织物起到保护作用,避免变黄、变硬,使颜色鲜艳并保持柔软。分离碱性纤维素酶基因、构建基因工程菌是提高碱性纤维素酶产量的重要方法之一[7-9]。此外,酶分子的定点突变和定向进化也为酶分子的改造提供了新的思路[10-13]。酶分子体外定向进化模拟自然进化,通过酶编码基因的一轮或多轮突变,构建酶突变基因文库,经过筛选得到所需性质的酶[14-15]。定向进化的方法主要有易错PCR、DNA shuffling等,根据待进化的酶分子特性可选择不同的方法。作者根据阳性转化子在IPTG诱导下可以在LB-CMC平板上产生水解圈的原理[16-17],在初筛和摇瓶复筛的基础上,采用易错PCR法对β-1,4-葡聚糖内切酶基因进行定向进化,从阳性转化子中筛选酶活提高的突变菌株。
1 实验
1.1 试剂与仪器
PCR产物纯化试剂盒、IPTG,上海生工;胶回收试剂盒,Qiagen公司;Taq DNA聚合酶、DNase Ⅰ、XhoⅠ、BamHⅠ限制性内切酶、T4 DNA连接酶、RNase A、dNTPs、DNA抽提试剂,大连宝生物公司。
热循环仪,凝胶成像系统,低温冷冻离心机,全温度恒温气浴摇床,电热恒温培养箱,酸度计,垂直净化工作台,电子天平,灭菌锅。
1.2 菌株、载体与培养基
大肠杆菌BL21(DE3)、表达载体pET20b,中科院昆明动物研究所;pMD18T,大连宝生物公司。
LB培养基[18]。
1.3 β-1,4-葡聚糖内切酶的易错PCR扩增
参照文献[18],提取短小芽孢杆菌DNA,克隆编码β-1,4-葡聚糖内切酶的基因,构建表达载体pET20b,转化大肠杆菌感受态细胞,得到重组菌BL21(DE3)/pET20b-EglA;在PCR反应体系中添加不同量Mn2+,进行易错PCR扩增,通过酶活比较筛选最佳的Mn2+添加量;参照TaKaRa公司的说明书进行易错PCR扩增产物DNA的酶切及连接;按Qiagen公司试剂盒说明进行易错PCR扩增产物DNA的回收。
1.4 易错PCR突变文库的构建及阳性转化子的筛选
设计一对引物5′-ATCTGGATCCATGCACATTTTTG-3′、5′-ATCGCTCGAGTTATTTATTCGGAAG-3′,分别引入BamHⅠ和XhoⅠ进行双酶切,对酶切片段进行鉴定[18],构建易错PCR突变文库。根据阳性转化子在IPTG诱导下可以在LB-CMC平板上产生水解圈的原理进行初筛,挑选水解圈与菌落直径比值较大的阳性转化子进行摇瓶复筛[19],以筛选出酶活提高的突变菌株。
1.5 β-1,4-葡聚糖内切酶重组菌的诱导表达及酶活测定
挑取BL21(DE3)/pET20b-Mut-EglA及BL21(DE3)/pET20b-EglA单菌落按1%的接种量接种至含50 μg·mL-1氨卞青霉素的LB液体培养基中,37 ℃振荡培养至OD600值在0.5~1.0之间;加入诱导物IPTG至终浓度为1 mmol·L-1,37 ℃诱导一定时间,取样测定酶活[19]。
1.6 突变酶与野生酶的表达量分析
将BL21(DE3)/pET20b-Mut-EglA和BL21(DE3)/pET20b-EglA分别于LB培养基中37 ℃培养10 h,按1%的接种量接种至LB液体培养基中,37 ℃培养至对数期,加入诱导物IPTG后继续培养4 h,10 000 r·min-1离心5 min,收集菌体,用2×SDS上样缓冲液于沸水浴中加热5 min裂解菌体,离心,取上清进行SDS-PAGE分析,利用BandScan软件测定突变酶及野生酶的表达量。
1.7 突变酶基因DNA及氨基酸序列分析
提取酶活提高突变菌株的重组质粒,进行DNA序列测定,在GenBank数据库中,利用BLAST软件对突变酶基因进行DNA及氨基酸序列分析。
2 结果与讨论
2.1 易错PCR碱基错配的引入
PCR反应体系中的Mg2+或Mn2+不仅会使Taq DNA聚合酶的保真性大大降低,还会影响酶活。Mn2+添加量对易错PCR产物的影响如图1所示。
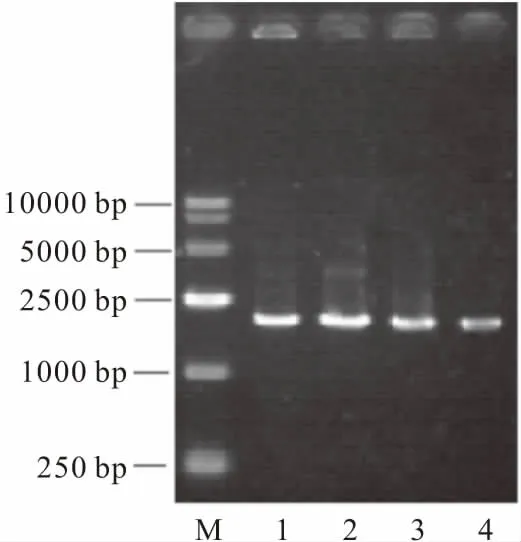
M.DL5000 DNA marker 1.Mn2+添加量1 mL 2.Mn2+添加量2 mL 3.Mn2+添加量3 mL 4.Mn2+添加量4 mL
从图1可以看出,随着PCR反应体系中Mn2+添加量的增加,DNA条带亮度减弱,说明催化合成的DNA量减少,突变率随着Mn2+添加量的增加而升高。
将添加不同量Mn2+扩增得到的PCR产物进行纯化、回收、酶切、连接表达载体,转化大肠杆菌,构建突变文库,根据平板上阳性转化子比率筛选合适的Mn2+添加量。结果表明,50 μL反应体系加入1 μL Mn2+,阳性转化子比率在60%左右,对建立随机突变体系较合适。若Mn2+添加量过多,反应体系的突变率太高,阳性转化子太少,此时大部分为负向突变,不利于筛选到正向突变菌株。
2.2 高酶活菌株的筛选
将易错PCR扩增得到的DNA片段构建到表达载体pET20b上,转化大肠杆菌,在IPTG诱导下,阳性转化子周围可以看到清晰的水解圈(图2a);挑选水解圈与菌落直径比值较大的阳性转化子进行摇瓶复筛(图2b),可以看到,大部分的阳性转化子都发生了负向突变,有些阳性转化子由于发生错配的碱基较多,从而失去了产酶活性,只有少部分的阳性转化子发生了正向突变。

图2 阳性转化子在LB-CMC平板上产生的水解圈
选取5株水解圈和菌落直径比值较大的阳性转化子,经过1轮易错PCR的定向进化,测得酶活(U·mL-1)分别为3.42、4.78、4.02、4.23、4.15,其中4株突变菌株所产酶的酶活得到提高,BL21(DE3)/pET20b-Mut-EglA2所产酶的酶活达到4.78 U·mL-1,为野生菌株BL21(DE3)/pET20b-EglA所产酶(3.60 U·mL-1)的1.32倍。
2.3 突变酶与野生酶的表达量分析
对BL21(DE3)/pET20b-Mut-EglA和BL21(DE3)/pET20b-EglA发酵产物进行SDS-PAGE分析,结果如图3所示。
从图3可以看出,诱导4 h后,菌株BL21(DE3)/pET20b-Mut-EglA和BL21(DE3)/pET20b-EglA均在73 kDa左右有β-1,4-葡聚糖内切酶目的蛋白的表达条带。利用BandScan软件测定蛋白条带的相对含量,发现突变酶的表达量比野生酶提高了5%,而酶活却提高了1.32倍,说明突变酶的催化效率得到了提高。
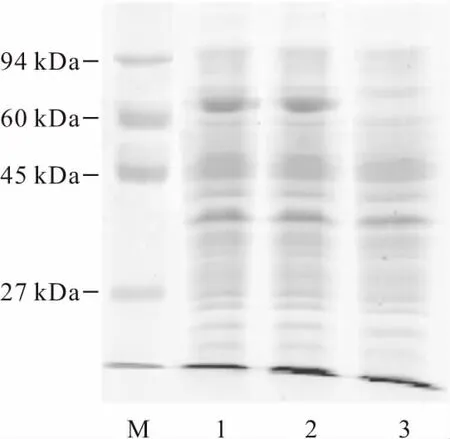
M.molecular mass marker 1.诱导4 h后的BL21(DE3)/pET20b-Mut-EglA 2.诱导4 h后的BL21(DE3)/pET20b-EglA 3.未诱导的BL21(DE3)/pET20b-EglA
2.5 突变酶与野生酶氨基酸序列的比较
基因测序结果表明,重组基因片段长度为1 980 bp。对DNA序列分析可知突变酶基因序列中有3个碱基发生了突变,其中2个为胸腺嘧啶碱基突变为胞嘧啶碱基,突变位点分别位于1 760 bp和1 962 bp处,导致酶的氨基酸序列中2个氨基酸发生了改变:其中野生酶突变位点的密码子AAA(106 bp)突变为GAA,其编码的氨基酸由赖氨酸变为谷氨酸;野生酶突变位点的密码子TTT(1 760 bp)突变为TCT,其编码的苯丙氨酸变为赖氨酸;而1 962 bp处密码子GGT突变为GGC,但是其编码的氨基酸没有发生改变,为同义突变。
2.6 讨论
应用PCR方法对酶分子进行改造,每一轮反应碱基突变太多会造成负向效应,一般要经过多轮反应才能将有益突变进行积累,使进化向正向突变定向进行,从而取得较好的效果。
一级序列差异很大的蛋白质能折叠成相似的三维结构,而仅有少数氨基酸差异的蛋白质却呈现不同的三维结构,蛋白质空间结构形成的规律还远未被认知,如何根据蛋白质的氨基酸序列折叠出其三维结构是当前研究的核心问题。突变酶氨基酸改变太少,其空间结构一般无法从三维结构上观察到。突变酶活性的提高可能是由于N端氨基酸的改变导致酶分子空间结构发生了改变,进而提高了其催化效率;也可能是由于C端结合结构域的氨基酸改变使空间结构发生了改变,导致酶分子周围底物浓度增大,从而使底物更容易与催化结构域结合,进而提高其催化效率[20-22]。
3 结论
根据阳性转化子在IPTG诱导下可以在LB-CMC平板上产生水解圈的原理,在初筛和摇瓶复筛的基础上,采用易错PCR法对β-1,4-葡聚糖内切酶基因进行定向进化,从阳性转化子中筛选酶活提高的突变菌株。突变酶活性是野生酶的1.32倍,催化效率约为野生酶的1.26倍。突变酶基因DNA序列中有3个碱基发生了突变,导致氨基酸序列中有2个氨基酸发生了改变:野生酶突变位点的密码子AAA(106 bp)突变为GAA,其编码的氨基酸由赖氨酸变为谷氨酸;野生酶突变位点的密码子TTT(1 760 bp)突变为TCT,其编码的苯丙氨酸变为赖氨酸;野生酶突变位点的密码子GGT(1 962 bp)突变为GGC,但是其编码的氨基酸没有发生改变,为同义突变。
