ATP7A基因c.1450G>T核苷酸变异致Menkes病1例并文献复习
2021-02-25徐德勇
徐德勇,陆 韦
(1.遵义医科大学,贵州遵义 563003; 2.遵义医科大学附属医院)
1 临床资料
患儿男,3个月,矫正胎龄2月余,因“发育落后半个月,抽搐1次”入院。半个月前,家属自觉患儿发育较同龄儿童落后,表现为不能追声、追物,反应差,头发不长,皮肤松弛,不能竖头,病初未予重视,今为进一步治疗就诊我院门诊。予完善血尿筛查,在等待过程中,患儿出现抽搐,表现为双目凝视,唇周微绀,口吐白沫,右上肢屈曲抽动,左上肢强直,双下肢抽动,无二便失禁,无烦躁不安,无发热。门诊立即予安定静推、吸氧、保持呼吸道通畅等对症处理,病情较前好转。患儿发病以来,精神、吃奶可,二便正常。既往无类似病史,否认家族史及头颅外伤史,系第1胎第1产,35+1周早产儿。以“(1)抽搐原因:癫痫?(2)全面发育落后原因:遗传代谢性疾病?”收住院。
入院查体:T 36.6℃,P 132次/分,R 34次/分,SPO297%(吸氧下),wt 6kg,头围39cm。神清,反应欠佳,不能抬头,头发少而黄,卷曲,鼻梁低平,眼距稍宽,眼裂小,追声、追物不能,呼吸规则;颜面、甲床及耳廓较苍白,全身皮肤松弛,弹性差,未见皮疹及出血点;双瞳孔正大等圆,对光反射灵敏,口唇无发绀,咽无充血,颈软;心肺腹查体未见明显异常,四肢肌张力减低,余神经系统查体未见异常(图1A、B)。
辅助检查:血常规:白细胞总数4.91×109/L,中性粒细胞百分比0.19,淋巴细胞百分比0.64,血红蛋白104g/L,血小板311×109/L,CRP正常。血生化:肝肾功、电解质、心肌酶及血糖未见异常。铜蓝蛋白2.59mg/dl,血清铜0.8μmol/L。头颅MRI:双侧大脑前、中、后动脉及基底动脉区、环池走形多发迂曲血管影,疑烟雾病可能(图1C)。头颅MRA:颅内多发迂曲血管影(图1D)。胸部CT提示:双肺肺炎,双侧肋骨形态不规则(图1E)。膝关节平片:双侧股骨下段干骺端增宽,侧棘形成,先期钙化带不规则,双侧胫腓骨近段干骺端稍增宽(图1F)。儿童脑电图:异常脑电图(双侧后头部尖波、尖慢波、棘慢波发放)。Gesell智能发育诊断量表:DQ=26.6,生长发育处于重度落后水平。脑干听力诱发电位:(1)双侧听神经远端或耳蜗异常可能,120dBpeSPL双侧I BAEP波潜伏期延长;(2)BAEP听阈异常增高;(3)耳声发射:双侧均未引出DPOAE。遗传代谢性指标未见异常。
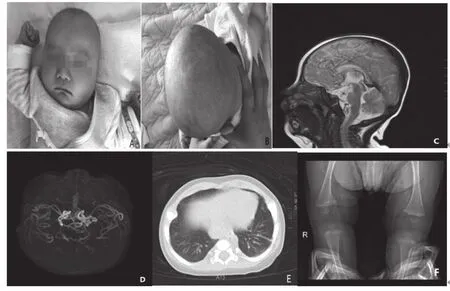
图1 患儿体貌特征及影像学检查结果
基因检测:北京康旭医学检验所进行家系全外显子组测序验证结果(Sanger测序验证),应用多重连接探针扩增技术检测ATP7A基因突变,在患儿ATP7A基因发现c.1450G>T的核苷酸变异,半合子变异,为无义变异,变异频率在ExAC、ESP6500、1000Genomes(CHB)、1000Genomes(CHS)、Kangso Health均未收录。ACMG评级(疑似致病)致病性报告:c.1450G>T变异的致病性尚未见文献报道(参考数据库:HGMD Pro、PubMed、ClinVar)。父亲未发现变异,母亲该位点为杂合子(见图2)。
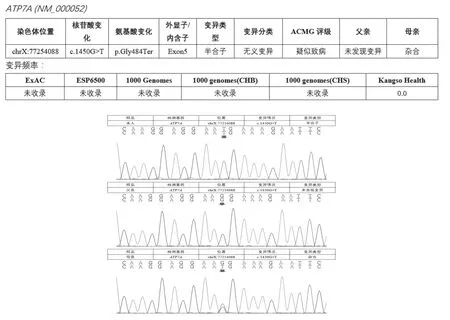
图2 患儿ATP7A基因测序图
诊断与治疗:临床拟诊Menkes病。住院期间,患儿喂养困难,竖头不稳,反复感染,多次出现抽搐,表现形式多样,持续时间约数秒至数分钟不等,予苯巴比妥片、托吡酯片抗癫痫治疗。因我院无组胺铜,家属进一步就诊于重庆医科大学附属儿童医院,并最终确诊为Menkes病。
2 讨论
Menkes病是由于ATP7A基因突变导致先天性铜代谢异常引起的进行性神经系统变性和结缔组织异常的X-连锁隐性遗传病[1]。ATP7A基因是一种编码膜功能蛋白,位于Xq21.1上的ATP酶家族,具有跨膜转运铜离子功能的离子泵,当ATP7A酶功能缺陷时,铜不能由肠黏膜上皮细胞转运至血液和其他组织中,导致血铜过低,诸多铜转运酶的缺陷导致了Menkes病临床特征的多样性,如酪氨酸酶影响毛发、皮肤色素的含量减少,细胞色素C氧化酶影响神经系统,抗坏血酸氧化酶影响骨骼发育[2]。ATP7A基因突变类型与Menkes病临床表型有关,经典型大部分发病在出生后2~3个月时,以反复癫痫发作为首发症状,易单纯诊断为癫痫,此型患儿预后差,通常死于3岁前,不能遗传下一代;而轻型及枕角综合征则多在青少年期或青少年早期发病,临床以结缔组织及骨骼改变为主,伴认知障碍,一般症状相对较轻。因为是X-连锁隐性遗传病,此病虽常见于男性,但也见于少部分女性,可遗传至下一代。轻型男性患者与正常女性生育的子女中,男性不发病,女性为携带者;轻型男性患者与携带者女性生育的子女中,男性50%发病,女性50%发病、50%为携带者;携带者女性与正常男性生育的子女中,50%的男性发病,50%的女性为携带者。约1/3的Menkes病患者没有家族史。
目前,对 Menkes病的诊断尚无金标准,主要是根据典型的临床特征和辅助检查、检测进行临床诊断,大部分患儿有早产病史,在生后的2~3个月即发病。主要临床特征:(1)癫痫频发,形式多样。(2)生长发育落后,表现为抬头、翻身、坐、走等发育停滞或缓慢,智力低下,肌张力减低,神经发育迟缓。(3)特殊面容,全身皮肤松弛、白皙、弹性差。(4)头发量少、短、色淡、卷曲、易碎,以两侧的颞部及枕后部为著。(5)可合并漏斗胸、脐疝、腹股沟疝。主要辅助检查表现为:(1)血清铜、铜蓝蛋白指标明显低于正常值。(2)显微镜下观察能发现结节性脆发或头发呈串珠样。(3)MRI表现为颅内血管迂曲,脑白质发育异常,脑萎缩,硬膜下积液或基底神经节受损等改变。(4)MRA 表现为颅内血管迂曲,“螺丝锥”样改变。(5)脑电图提示异常脑电图改变。(6)X线表现为长骨干骺端骨刺形成,呈“杯口”样改变,胸骨可见不规则膨大。(7)听觉及视觉诱发电位提示振幅减低或消失[3-8]。其中颅内血管迂曲是Menkes最为突出的表现之一,具有影像学诊断意义,确诊还需依赖于分子遗传学检测[9]。
婴儿期有明显的运动发育落后、频繁的癫痫发作,而且为早产儿,应考虑可能为遗传代谢病。本例患儿父亲未发现变异,变异来源于母亲,其母为杂合子,可能为母亲基因突变所导致,符合X连锁隐性遗传方式。故为有效降低Menkes病患儿的出生,需行产前诊断及遗传咨询[10-11]。
我们在万方、中国知网和PubMed数据库中检索该病,检索时间为2016~2020年,国内有51篇相关文献报道,国外有70篇相关文献报道。ATP7A基因变异已经收录达354种之多,主要包括错义突变、无义突变、剪切变异、小片段的缺失或插入和大片段缺失或插入[11]。其中最新发现的致病性变异主要有:错义突变(c.3028A>C),无义突变(c.1642G>T;c.2922C>A),剪切突变(IVS18+1G>A; IVS10+1G>A),小片段缺失或插入(c.2589_2612delinsCACAG;c.1642_1643insA;c.1027_1028insT;IVS20-7_IVS20-3delTTCTT),大片段缺失(IVS9-346_IVS10 + 1203del;c.1-?_1708 + ?del;c.3112-? + 3112 + ?del)[12]。
ATP7A基因c.1450G>T的核苷酸变异是我们首例发现的突变位点,属于无义突变,该突变位点此前尚无报道。因此,该突变位点的发现,丰富了Menkes病的基因谱,为开展产前诊断及遗传咨询、减少Menkes病患儿的出生提供了分子生物学基础,也为Menkes病的诊断提供了临床依据。
