Single-cell RNA Sequencing Reveals Thoracolumbar Vertebra Heterogeneity and Rib-genesis in Pigs
2021-02-24JianboLiLigangWangDaweiYuJunfengHaoLongchaoZhangAdeniyiAdeolaBingyuMaoYunGaoShifangWuChunlingZhuYongqingZhangJilongRenChanggaiMuDavidIrwinLixianWangTangHaiHaibingXieYapingZhang
Jianbo Li, Ligang Wang, Dawei Yu, Junfeng Hao, Longchao Zhang,Adeniyi C.Adeola,Bingyu Mao,7,Yun Gao,Shifang Wu,Chunling Zhu,Yongqing Zhang, Jilong Ren, Changgai Mu,6, David M. Irwin,7, Lixian Wang, Tang Hai,,Haibing Xie,, Yaping Zhang,6,8,
1State Key Laboratory of Genetic Resources and Evolution, Yunnan Laboratory of Molecular Biology of Domestic Animals, Kunming Institute of Zoology, Chinese Academy of Sciences, Kunming 650223, China
2Institute of Animal Science, Chinese Academy of Agricultural Sciences, Beijing 100193, China
3State Key Laboratory of Stem Cell and Reproductive Biology,Institute of Zoology,Chinese Academy of Sciences,Beijing 100101,China
4Core Facility for Protein Research, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China
5State Key Laboratory for Molecular and Developmental Biology, CAS Center for Excellence in Brain Science and Intelligence Technology, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, Beijing 100101, China
6Kunming College of Life Science, University of Chinese Academy of Sciences, Kunming 650223, China
7Department of Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario M5S1A8, Canada
8Center for Excellence in Animal Evolution and Genetics, Chinese Academy of Sciences, Kunming 650223, China
Abstract Development of thoracolumbar vertebra (TLV) and rib primordium (RP) is a common evolutionary feature across vertebrates,although whole-organism analysis of the expression dynamics of TLV-and RP-related genes has been lacking.Here,we investigated the single-cell transcriptome landscape of thoracic vertebra(TV),lumbar vertebra(LV),and RP cells from a pig embryo at 27 days post-fertilization (dpf) and identified six cell types with distinct gene expression signatures. In-depth dissection of the gene expression dynamics and RNA velocity revealed a coupled process of osteogenesis and angiogenesis during TLV and RP development. Further analysis of cell type-specific and strand-specific expression uncovered the extremely high level of HOXA10 3′-UTR sequence specific to osteoblasts of LV cells,which may function as anti-HOXA10-antisense by counteracting the HOXA10-antisense effect to determine TLV transition.Thus,this work provides a valuable resource for understanding embryonic osteogenesis and angiogenesis underlying vertebrate TLV and RP development at the cell type-specific resolution, which serves as a comprehensive view on the transcriptional profile of animal embryo development.
KEYWORDS scRNA-seq;Thoracolumbar vertebra transition;Rib-genesis;Osteogenesis;Angiogenesis
Introduction
In vertebrates,vertebrae develop and segment during early embryogenesis [1]. During this process, cervical vertebra(CV), thoracic vertebra (TV), lumbar vertebra (LV), sacral vertebra (SV), and caudal vertebra (CAV) are formed in sequence along the anterior–posterior axis [2,3]. Body region allocation and the transition between regions have important morphological, physiological, and evolutionary consequences, given that their relative proportions vary widely among vertebrates[4].Partitioning of the body into TV and LV has been of long-term biological research interest, and many pioneering studies have attempted to identify genes and genomic variations underlying this developmental process. For example,Oct4andGdf11are identified as important genes, and their overexpression or knockout in mice leads to TVelongation,similar to the long TV partition observed in snakes[5,6].At the same time,the thoracolumbar vertebra (TLV) transition is shaped by members of theHoxgene cluster in mice[7–9].Therefore,despite the valuable insights into TLV transition and ribgenesis at the single-gene level, previous transcriptomic analyses have not resolved the tempo-spatial gene expression patterns underlying these developmental processes.Thus, profiling of gene expression in TV and LV body partitions offers an opportunity to gain deeper insights into these developmental processes.
The development of single-cell RNA sequencing(scRNA-seq) technologies has provided an opportunity to investigate tempo-spatial gene expression during embryo development. scRNA-seq methods have higher gene expression resolution than traditional transcriptome analyses,such as whole-embryo transcriptome sequencing and bulk RNA sequencing [10,11]. scRNA-seq has an advantage in detecting cell types and gene expression signatures for each type [12,13]. For instance, cell atlases for mammalian systems, including neonatal rib and bone marrow stroma,have been generated by analyzing both fetal and adult mouse tissues[14–16].Cell atlas characterization and gene expression analysis would provide valuable information on the difference between TV and LV development.
Previous studies on TVand LV development have largely focused on mouse models by examining the effects of genetic variation on phenotypes through the overexpression or knockout of genes [7–9]. These models have greatly advanced our knowledge on TV and LV partition development. Alternative models for studying TV and LV development include some domestic animals with varying numbers of TV and LV, such as pigs and sheep [17,18].These domestic animals may offer valuable model species for further exploration of genes and signaling pathways involved in TV and LV development with a low genomic divergence among individuals within a species.
In this study,we used the pigs as a model to explore the cell compositions in the developing TV, LV, and rib primordium (RP). We conducted a single-cell transcriptome analysis of cells collected from TV, LV, and RP from one large white (LW) pig embryo at 27 days post-fertilization(dpf), which corresponds to the commencement of rib formation.Overall,this study provides a rich resource that can advance our understanding of TLV transition and RP development in vertebrates.
Results
Cell composition and differentiation trajectory of developing TLV
To gain an insight into the development of TV and LV, we started an analysis by characterizing cell populations from the two different anatomical body partitions. To determine the time point for cell sampling, we examined the development of pig embryos at 20,25,27,and 29 dpf.Our analysis revealed that ribs commenced stemming from TV at 27 dpf, while embryos less than 27 dpf did not show evident ribcages. RP development completed at 29 dpf(unpublished data). Therefore, the embryo at 27 dpf was used for cell population characterization in developing TV and LV.
A total of 360 cells(180 TV cells and 180 LV cells)from six consecutive vertebrae(three TVand three LV segments)close to the TLV segmentation joint were isolated by micromanipulation and enzyme digestion from an LW pig embryo at 27 dpf (Figure 1A, Figure S1). We performed Smart-seq2 for full-length transcriptome profiling on the TV and LV cells. After stringent filtration, 265 cells (128 TV cells and 137 LV cells) were retained for further analyses. The TV and LV cells were integrated and classified into six clusters (clusters 1–6) using the function‘FindCluster’in Seurat,while no distinct cluster was found between TV and LV(Figure 1B).To identify the properties of different cell clusters, we identified and analyzed differentially expressed genes (DEGs) in each cluster, including previously identified cell type-specific markers in each cluster. As shown in Figure 1C, cluster 1 specifically expressedCOL1A1[19] andEBF2[20], as well as osteoblast (OB) development-related genes, such asOGN[21]andGAS2[22], and thus was classified as OB. Cluster 2 specifically expressed fibroblast (FB) development-related genesLUM[23],DCN[24], andTCF4[25], and thus was classified as FB. Cluster 3 expressedHMGB2, as well as cell mitosis-related genes,such asTOP2A,MXD3,CDCA3,CDC20, andCKAP2. AsHMGB2plays a role in osteogenesis [26] and mesenchymal stem cell (MSC)differentiation[27],this cell cluster was classified as stroma cell (SC). Cluster 4 specifically expressedMATN1,COL11A1,COL11A2,MATN4,andMATN3[28],as well as cartilage (CT) development-related genes, such asCNMD,EPYC,HAPLN1, andPCOLCE2[29], and thus was classified as CT. Cluster 5 specifically expressedCD248[30]andBMP4[31], as well as MSC differentiation-related genes, such asASB9,MAB21L2,SERPINF1,NNAT, andCLDN11, and thus was classified as MSC. Cluster 6 specifically expressedCD34[32,33]andCD93[34],as well as angiogenesis-related genes,such asPRCP[35]andEMCN[36],and thus was classified as hemogenic endothelial cell(HEC).
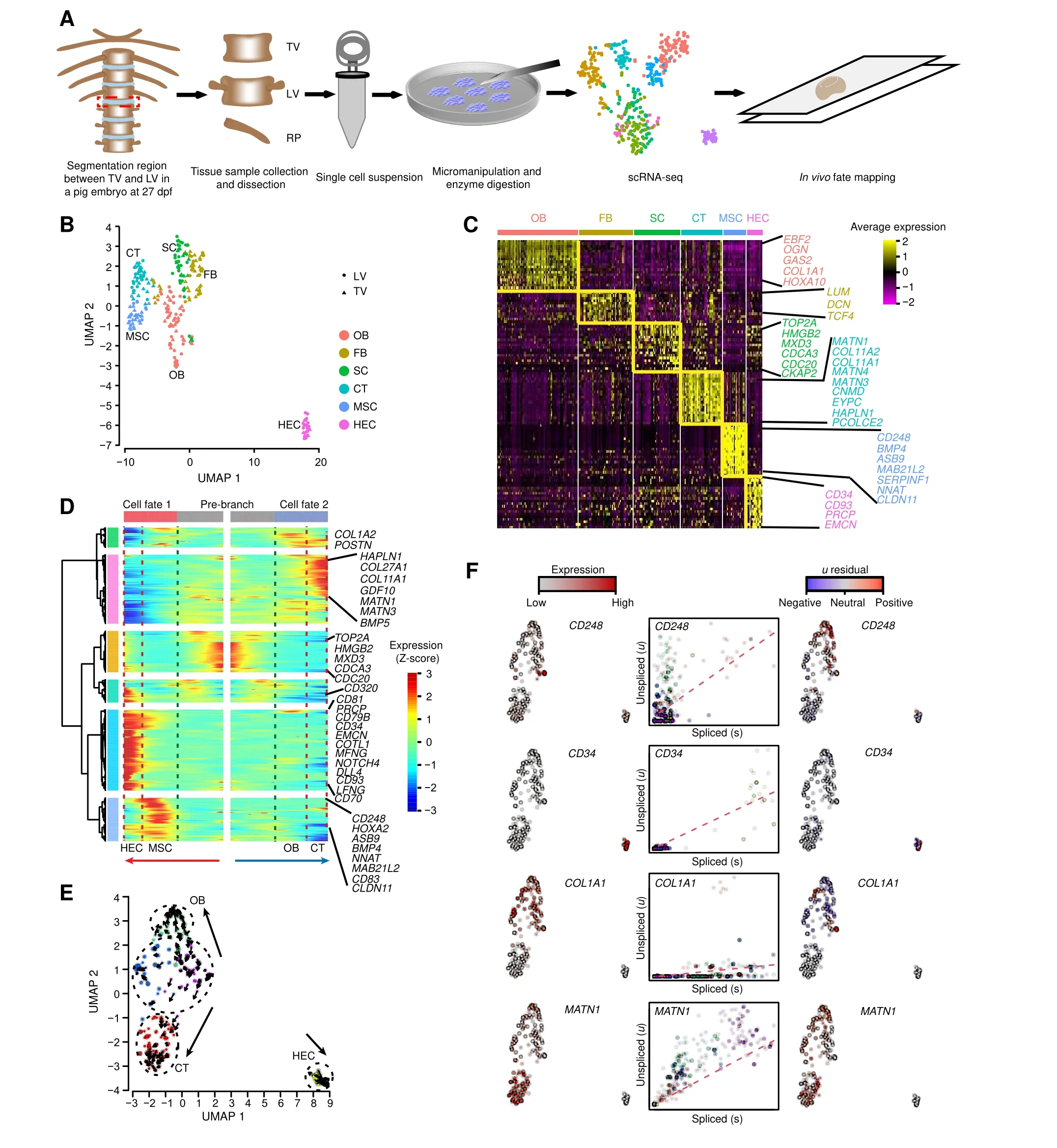
Figure 1 Cell composition and differentiation trajectory of developing TLVA.Overview of the experimental design.Segmentation regions between TV and LV were dissected in pigs and dissociated into single cell suspensions by micromanipulation and enzyme digestion.Red dashed rectangle indicates the TLV segmentation joint.Smart-seq2 was used for scRNA-seq.B.Integrated UMAP plot of cell clusters from TV and LV. C. Gene expression patterns of each cell cluster in(B). Cell type-enriched genes are listed on right and are labeled in the same colors as corresponding cell types. D. Bifurcation of the 799 top TLV DEGs along two branches clustered hierarchically into six modules in a pseudo-temporal order. Development trajectories of TV and LV cells are shown on the right and left, respectively. Red arrow indicates the pseudo-time of cell fate 1(MSC to HEC);blue arrow indicates the pseudo-time of of cell fate 2(OB to CT).Representative genes are shown on the right.E.RNA velocity recapitulating the dynamics of TLV cell differentiation.The arrows indicate the position of the future state.F.Expression pattern(left),unspliced–spliced phase portrait (middle; cells colored according to E), and u residual (right) of TLV cells are shown for CD248, CD34, COL1A1, and MATN1. TLV, Thoracolumbar vertebra; TV, thoracic vertebra; LV, lumbar vertebra; scRNA-seq, single-cell RNA sequencing; UMAP, uniform manifold approximation and projection;OB,osteoblast;FB,fibroblast;SC,stroma cell;CT,cartilage;MSC,mesenchymal stem cell;HEC,hemogenic endothelial cell; DEG, differentially expressed gene.
To reconstruct the developmental processes of TV and LV cells, we performed Monocle-derived pseudo-time analysis.The TVand LV cells were successfully distributed along pseudo-temporal paths consisting of a pre-branch and two cell fates: MSC–HEC (cell fate 1; angiogenesis) and OB–CT (cell fate 2; osteogenesis) (Figure 1D). The angiogenesis branch was consistent with observations from previous reports suggesting that MSC could be differentiated into endothelial cellsin vitroandin vivo[37,38]. These paths harbored cell type-specific markers,includingCOL1A2[39],MATN1[40],CD34[32,33], andCD248[30]. RNA velocity analysis further confirmed the general pattern of TLV cell differentiation associated with osteogenesis and angiogenesis (Figure 1E). The prediction of transcriptional dynamics of the developing pig TLV cells showed that OB and CT are from MSC, constituting the largest branch of differentiating lineages of the TLV cells.Our analysis revealed the expression of many marker genes,replicating the observation of expression ofCD248[30]in the MSC zone,COL1A1[19]in the OB zone,MATN1[40]in the CT zone, andCD34[32,33] in the HEC zone(Figure 1F).
Cell composition and HOXA10 expression difference between developing TV and LV
To explore cell composition difference, we compared the fractions of cell populations in devopling TV and LV. Cell cluster analysis revealed that both TVand LV contained six clusters of cells (Figure S2), consistent with results observed in the whole-cell samples(Figure 1B);however,the fractions of cells in some clusters differred between TVand LV. The highest fractions were in the CT cell cluster from both groups, but showed no significant fraction difference(Permutation test,n= 1,000,000 replicates;P= 0.51). For the FB,SC,HEC,and MSC cell clusters,fraction difference was statistically significant when cell clusters from TV and LV were compared(FB,P=1.16×10−2;SC,P=2.25×10−2;HEC,P= 7 × 10−2; MSC,P= 5.04 × 10−3). Moreover, no fraction difference was observed in the OB cell clusters from TV and LV (P= 0.15).
Next,we compared the top 20 highly expressed genes in the same cell clusters from TVand LV.Results showed that most of the genes were shared by the same cell clusters from TV and LV;however,there was a difference in the order of expression level in TV and LV(Table S1).Interestingly,we found thatHOXA10showed differential gene expression in the OB cell cluster from TV and LV. We observed thatHOXA10was the top gene with the highest expression level in OB from LV, but was nearly absent in OB from TV.Additionally,HOXA10was not expressed in most TV and RP cells[118 of 128 TV cells and 174 of 199 RP cells,with reads per kilobase of exon model per million mapped reads(FPKM)<1],whileHOXA10had a high expression level in most LV cells(79 of 137 LV cells,with FPKM>5).Further validation using all of the single-cell transcriptome data showed that the expression ofHOXA10was largely restricted to cell clusters from LV,not in cell clusters from TV(Figure 2A).In the comparison of gene expression levels in OB cell clusters from TV and LV,HOXA10showed the largest expression bias toward LV (Figure 2B). Moreover,HOXA10showed a wide expression bias toward cell clusters from LV,in comparison to cell clusters from TV(Figure 2C). A similar but incomplete pattern was observed forHOXC10,but not forHOXD10(Figure 2C).Taten together,these results indicate thatHOXA10may function as a determining factor that separates the sampled cells into either the TV or LV lineage.
To further characterize the expression ofHOXA10in developing TV and LV in pig embryo, we compared the distribution of sequencing reads at this locus (Figure 2D).On average, the sequencing depth of the reads in theHOXA10coding sequence was about 47.87×in LV cells and only 0.24×in TV cells.Unexpectedly,the sequencing depth in the 3′-UTR ofHOXA10in both cells from LV(1547.88×)and TV(70.57×)was at least 30-fold higher than that for the coding sequence.An analysis of the gene structure showed that an antisense RNA,HOXA10-AS, overlaps the 3′-UTR on the opposite strand of theHOXA10gene. A possible explanation for the observed higher sequencing depth at the 3′-UTR is either a higher level ofHOXA10-AS expression orHOXA103′-UTR expression, since scRNA-seq cannot distinguish strand-specific RNA expression. Nevertheless,the average FPKM ofHOXA10-exon3 was extremely lower than those forHOXA10-AS among the 137 LV cells(Figure 2E), with 0.18 forHOXA10-exon3 and 6.58 forHOXA10-AS(P=1.056E−07,unpaired two-sided Welch’st-test). To estimate the contribution ofHOXA10andHOXA10-AS expression to theHOXA103′-UTR genomic region, strand-specific expression was quantified using reads containing poly(A) tail (Figure 2F). Among the 137 LV cells,HOXA10poly(A) tail were detected in 44 cells,whileHOXA10-AS poly(A) tail were detected only in 13 cells.In addition,the number of reads containingHOXA10poly(A) tail was also much higher than that containingHOXA10-AS poly(A) tail in the 44 LV cells, with an estimate of ten-fold of the number (Figure 2F). This implies that the high sequencing depth from theHOXA103′-UTR genomic region was mainly due toHOXA10expression,rather thanHOXA10-AS expression. We failed to find any reads containingHOXA10poly(A)tail orHOXA10-AS poly(A) tail in the 128 TV cells, possibly due to the low expression level of these loci.

Figure 2 Cell heterogeneity and HOXA10 expression difference between developing TV and LVA.Median scaled ln-normalized gene expression of selected DEGs for LV and TV cell clusters.B.Scatter plot comparing the average expression levels of genes in OB cell clusters from LV and TV.C.Vinplot comparing expression of HOXA10,HOXC10,and HOXD10 in each cell cluster from LV and TV.D.Average sequencing depth of HOXA10 coding sequence, HOXA10 3′-UTR, and HOXA10-AS in 137 LV and 128 TV cells. Shade rectangles indicate the region of HOXA10 coding sequence and HOXA10 3′-UTR.E.Boxplot comparing the FPKM values of HOXA10-exon3 and HOXA10-AS in 137 LV cells.P value was obtained by unpaired two-sided Welch’s t-test with correction for multiple comparisons.F.Scatter plot showing the number of reads harboring HOXA10 poly(A) tail and HOXA10-AS poly(A) tail at the HOXA10 3′-UTR locus in 137 LV cells. HOXA10-AS indicates an antisense RNA which overlaps the 3′-UTR on the opposite strand of the HOXA10 gene. FPKM, reads per kilobase of exon model per million mapped reads.
Cell composition and differentiation trajectory of developing RP
There is insufficient knowledge on the gene expression profile involved in RP development in vertebrates. To understand this process,we collected 400 RP single cells from three consecutive TV at the TLV segmentation joint for Smart-seq2 transcriptome profiling. After stringent filtration and classification using the function ‘FindVariableFeatures’and‘FindCluster’in Seurat,199 RP cells were retained and classified into six clusters (clusters 1–6)(Figure 3A and B). Cluster 1 specifically expressed FB development-related genesTBX3[41],ASPN[42],YAP1[43], andSEMA3A[44], and thus was classified as FB.Cluster 2 specifically expressedMATN1,COL11A1,COL11A2,MATN4, andMATN3[28,40], and thus was classified as CT. Cluster 3 specifically expressedHMGB2,TOP2A,MXD3,CDCA3,CDC20, andCKAP2, and thus was classified as SC [26,27]. Cluster 4 specifically expressedEBF2,OGN,COL3A1, andCOL1A1[19,39], and thus was classified as OB.Cluster 5 specifically expressedBMP4,FOS,FOSB,GADD45B, andCD248[30,31], and thus was classified as MSC.Cluster 6 specifically expressedLAPTM5,PRCP,COTL1,CD93, andCD34[32–35], and thus was classified HEC.
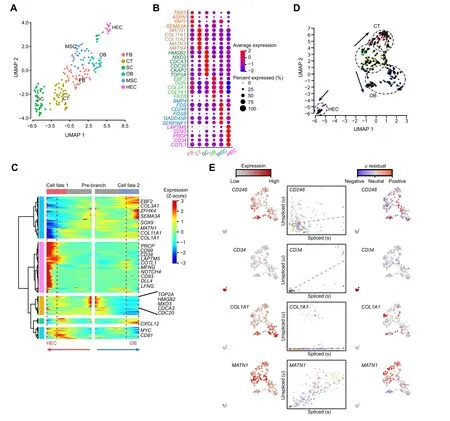
Figure 3 Cell composition and differentiation trajectory of developing RPA.Integrated UMAP plot of cell clusters from RP.B.Median scaled ln-normalized gene expression of selected DEGs for RP cell clusters from(A).Cell type-enriched genes are listed on the right and labeled in the same colors as corresponding cell types.C. Bifurcation of the 381 top RP DEGs along two branches clustered hierarchically into five modules in a pseudo-temporal order. Development trajectories of RP cells are shown on the right and left,respectively.Red arrow indicates the pseudo-time of cell fate 1(HEC);blue arrow indicates the pseudo-time of cell fate 2(OB).Representative genes are shown on the right. D. RNA velocity recapitulating the dynamics of the RP cell differentiation. The arrows indicate the position of the future state. E.Expression pattern (left), unspliced–spliced phase portrait (middle; cells colored according to D), and u residual (right) of the RP cells are shown for CD248, CD34, COL1A1, and MATN1. RP, rib primordium.
Further, we performed Monocle-derived pseudo-time analysis to reconstruct the RP developmental process. RP cells were distributed along pseudo-temporal paths consisting of a pre-branch and two cell fates:HEC(cell fate 1;angiogenesis) and OB (cell fate 2; osteogenesis) (Figure 3C). These paths harbored cell type-specific markers, includingCD34[32,33],CD93[34],EBF2[20],COL1A1[19], andSOX9[45], consistent with the gene expression patterns seen in Figure 3B. Similar results were obtained from RNAvelocity analysis that predicted the transcriptional dynamics of the developing pig RP cells(Figure 3D and E),indicating robustness for the classification of angiogenesis and osteogenesis during the RP developmental process.
Osteogenesis network construction and cell typespecific marker immunofluorescence analysis of developing TLV and RP
To reveal the features of osteogenesis during TLV and RP development in pigs, we conducted a weighted gene co-expression network analysis (WGCNA) to construct a gene correlation network. As a CT marker and top transcribed gene in the CT cluster,MATN1and its correlated genes were selected to build an osteogenesis network.MATN1has been identified as a vital gene for CT networks in humans and mice[28,40].Here,the hub genes correlated withMATN1includedCOL11A1,COL2A1,CNMD,EPYC,COL11A2,PCOLCE2,andHAPLN1,most of which are key genes involved in bone formation and remodeling [46](Figure 4A).
To confirm the spatial relationships among osteogenic cell types identified by Smart-seq2, we performed immunofluorescence imaging of TV and LV sections using a pig embryo at 27 dpf.MATN1[40],COL1A1[19], andHMGB2[26,27],which have been identified as markers for CT,OB,and SC,respectively,were selected based on Gene Ontology (GO) analysis of DEGs, and their relevant proteins were selected for immunofluorescence analysis. Signals for the three proteins were detected in TV,LV,and RP(Figure 4B–E). MATN1, as a secreted protein, was also detected inside TV, LV, and RP in our current study. In addition, HMGB2 was mainly detected in the nuclei and had a higher expression level in LV than in RP.COL1A1,as a secreted protein,was detected at the edges of TV,LV,and RP.In terms of osteogenesis,our data imply that COL1A1 is first expressed at the edge of neonatal bone to generate OB,and then MATN1 is rapidly activated inside the neonatal bone to remodel and form CT during TLV and RP development in a pig embryo at 27 dpf.
Angiogenesis network construction and cell typespecific marker immunofluorescence analysis of developing TLV and RP
Previous studies have shown that angiogenesis and osteogenesis are coupled in a specific vessel subtype during bone formation[36,45],while the features of angiogenesis during TLV and RP development remain unclear. As an HEC marker and top transcribed gene in the HEC cluster,CD34and its related genes were selected to build an angiogenesis network by WGCNA [32,33]. The hub genes correlated withCD34[32,33] includedCD93[34],PECAM1(also knownCD31) [36],PLVAP[47],EMCE[36],F11R(also knownCD321) [48],NPR1[49], andPRCP[35], most of which are involved in angiogenesis (Figure 5A). These results indicated thatCD34andCD93are two putative coordinators in the angiogenesis genetic network during early angiogenesis of TLVand RP development,in addition toPECAM1andEMCN[36]. Furthermore, pseudotemporal order analyses of both TLV and RP based on the top DEGs suggested the involvement of Notch pathway components in angiogenesis, includingDLL4,MFNG,LFNG,andNOTCH4(Figures 1D and 3C),consistent with previous reports suggesting that endothelial NOTCH activity promotes angiogenesis and osteogenesis in bone formation[50].These results reconfirmed that cluster 6 of TLV and RP cells is HEC rather than a hematopoietic stem cell.
To confirm the spatial relationships among the angiogenesis cell types identified by Smart-seq2, immunofluorescence imaging of TV and LV sections were performed using a pig embryo at 27 dpf.CD34[32,33],CD93[34], andCD248[30], which were identified as markers of HEC or MSC, were selected based on GO analysis of DEGs,and their relevant proteins were selected for immunofluorescence analysis. Signals of the three proteins were detected in TV, LV, and RP, as well as their surrounding tissues(Figure 5B and C).CD34,as a secreted protein, was highly expressed in the notochord and tissues surrounding TV, LV, and RP but was relatively less in TV,LV,and RP.CD93 was expressed at a high level at the edge of RP, as well as in TV, LV, and RP. In contrast, CD248,which is a membrane protein,was expressed at a high level at the edges of TV, LV, and RP, but with almost no expression in TV,LV,and RP,implying the synergistic action between MSC and OB during the skeletal system development and remodeling. These data indicate that angiogenesis and osteogenesis are coupled by specific HEC during TLV and RP development in pig embryos at 27 dpf.
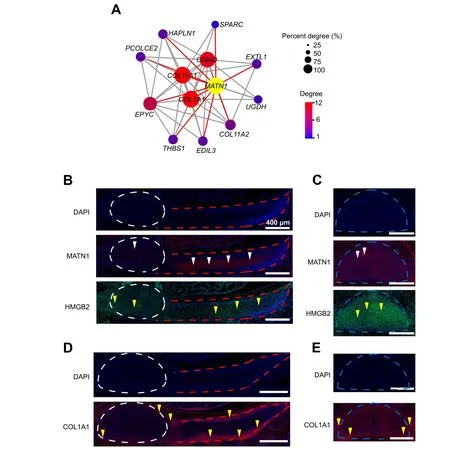
Figure 4 Osteogenesis network construction and cell type-specific marker immunofluorescence analysis of developing TLV and RPA. Module visualization of the network connections and associated functions using MATN1 as a hub gene. Gene-connected intra-modular degree is simultaneously indicated by spot size and color intensity.The hub gene MATN1 is indicated in yellow.B.and C.Immunofluorescence analysis of MATN1 and HMGB2 in TV and RP (B) and in LV (C). Red and green indicate fluorescence signals of MATN1 and HMGB2, respectively. White and yellow triangles indicate MATN1+ and HMGB2+ cells, respectively. D. and E. Immunofluorescence analysis of COL1A1 in TV and RP (D) and in LV (E). Red indicates fluorescence signals of COL1A1.Yellow triangles indicate COL1A1+cells.White,blue,and red dashed lines in(B–E)indicate regions of TV,LV,and RP, respectively. Scale bar, 400 μm.

Figure 5 Angiogenesis network construction and cell type-specific marker immunofluorescence analysis of developing TLV and RPA. Module visualization of the network connections and associated functions using CD34 as a hub gene. Gene-connected intra-modular degree is simultaneously indicated by spot size and color intensity. The hub gene CD34 is indicated in yellow. B. and C. Immunofluorescence analysis of CD93,CD248,and CD34 in TV and RP(B)and in LV(C).Green,white,and red indicate fluorescence signals of CD93,CD248,and CD34,respectively.White,yellow,and blue triangles indicate CD93+,CD248+,and CD34+cells.White,blue,and red dashed lines in(B and C)indicate the regions of TV,LV,and RP,respectively. Scale bar, 400 μm.
Discussion
In this study,we demonstrated that the domestic pig can be a valuable animal model for exploring the molecular mechanisms underlying TLV transition and RP development in vertebrates. Despite the inter-specific difference of embryonic developmental processes,the overlap of the DEGs observed in pigs and those reported in earlier mouse models implies a conserved regulatory feature of TLV and RP development among species [19,32,36]. The domestic pig may have advantages in exploring the genetic mechanisms underlying the TLV transition,since the number of TLV in different domestic pigs varies [17]. The intra-specific developmental variation may offer an opportunity for future studies to explore the genomic coordination that determines the TV and LV body identities, giving a low level of genomic noise within a single species.The results obtained in this study open a new window and provide valuable resources to expand such studies on TLV transition using pigs.
Our analysis revealed that the cell types in developing TLV can be functionally clustered into two groups, corresponding to the osteogenesis (OB–CT) and angiogenesis(MSC–HEC) biological processes. RP cells were functionally correlated to osteogenesis (OB) and angiogenesis(HEC). Our observations revealed a coupled process of osteogenesis and angiogenesis during TLV and RP development, highly consistent with observations from previous studies during bone formation [36,50]. This implies that the number of sampled cells may have substantial information to represent cell atlases of developing TLV and RP. This study may allow the discovery of transcriptome kinetics at the temporal resolution using scRNA-seq data and provides fundamental insights relevant to abnormal TLV transition and RP development in vertebrates. Our results are based on a limit number of embryos and cells.Further analysis using more embryos and cells is required in future studies.
Previous studies via intervention of gene expression,whole-mountin situhybridization, and immunofluorescence analysis revealed a crucial role ofHOXA10in TLV transition, while the molecular mechanism howHOXA10determines TLV transition remains elusive[7–9].In this study, we discovered the extremely high level ofHOXA10restricted to OB of LV cells rather than in TV cells using our scRNA-seq data. In-depth dissection of the read distribution revealed that most reads were restricted to the 3′-UTR ofHOXA10(overlapping withHOXA10-AS)rather than the coding region.HOXA10-AS is capable of repressingHOXA10expression [51,52]. The strand-specific expression analysis using reads containing poly(A) tail revealed that the high read sequencing depth fromHOXA103′-UTR genomic region was mainly due toHOXA10expression, rather thanHOXA10-AS expression. These observations suggested that TLV transition involves a putative expression balance between theHOXA10andHOXA10-AS genes.The large amount of reads clustered in theHOXA103′-UTR genomic region indicates the presence of a regulatory mechanism that blocks the expression ofHOXA10-AS through the expression of a short transcript from the 3′-end of theHOXA10sense strand that is complementary to theHOXA10-AS sequence, implying an anti-HOXA10-AS role of the high levels ofHOXA103′-UTR sequence within OB of LV cells.
It has been clearly shown that HECs are from the aortagonad-mesonephros region where hematopoiesis takes place [53,54]. Previous studies on hematopoiesis have focused on the liver and bone marrow,but insufficiently in somite,TLV,and RP[55–57].In zebrafish,somatic cells in embryos have been shown totrans-differentiate into hematopoietic cells by transgenic lineage tracing, suggesting that somite is an additional embryonic hematopoietic site[58,59]. Our results revealed a specific vessel subtype,HEC, with distinct molecular and functional properties during TLVand RP development in a pig embryo at 27 dpf.The angiogenesis gene network was established as early as four weeks post-fertilization in TLV and RP of pigs.
The bone developmental process includes four stages:pre-CT stage (spongy bone), CT stage, CT erosion (cancellous bone), and bone deposition (compact bone) [60].Here, we focused on the stage during which spongy bone turns into CT, but found no notable difference in the cell clusters between TV and LV.Sampling at 17 to 27 dpf may provide insights on these possible differences; however, it would be difficult to confirm the TLV segmentation joint,because RP is not developed in pigs before 27 dpf (unpublished data).
In this study, we compared six consecutive segments from TV and LV close to TLV segmentation joint from a single LW embryo, rather than to compare segments at the TV–LV boundary from different embryos. It was largely due to the challenge in sampling cells using micromanipulation in developing embryos and the high cost of scRNA-seq.Despite the lack of information from different embryos, the high consistency between our observations and earlier reports on the role ofHOXA10expression in discriminating TV and LV segments indicates the high confidence of the observations in this study, possibly because these cells substantially represent the TV and LV partitions. Analysis with more embryos is required to explore the cell compositions and expression features of TV,LV,and RP segments,as well as the difference between TV and LV in future studies.
In summary, our comprehensive atlases of TLV and RP from a pig embryo at 27 dpf provide a valuable resource for understanding molecular programs and development temporary states during TLV transition and rib-genesis in vertebrates. Our approach using single-cell transcriptomics to study TLV and RP development in pigs provides a framework that could be applied to study temporal processes in other animal models.
Materials and methods
Sample collection and preparation of single cell suspensions
TV,LV,and RP cells from six consecutive vertebrae(three TV and three LV segments)close to the TLV segmentation joint of one LW pig embryo at 27 dpf were collected by micromanipulation and enzyme digestion. TV, LV, and RP cells were uniformly dissected into millimeter-sized pieces in Dulbecco’s phosphate-buffered saline (DPBS; Catalog No. 14190136, Gibco, Carlsbad, CA) supplemented with 10%fetal calf serum,transferred to tubes containing 1 ml of collagenase II(5 mg/ml;Catalog No.C5138-1G,Sigma,St.Louis, MI) and 1 ml of dispase (2.5 mg/ml; Catalog No.42613-33-2, Sigma), and then incubated at 37°C for 3–5 min.Digested tissue pieces were then filtered through a 40-mm nylon cell strainer(Catalog No.352340,BD Falcon,Franklin,NJ)and centrifuged at 500gfor 10 min at 37°C to collect cell pellets.The cell pellets were next washed using 1× DPBS three times to remove fragments and then resuspended in Dulbecco’s Modified Eagle’s Medium (Catalog No. 11995040,Gibco).
Full-length scRNA-seq library preparation and sequencing
We used the Smart-seq2 protocol for full-length scRNA-seq according to the manufacturer’s instructions [61]. Briefly,single cell was transferred to lysis buffer with RNase inhibitor in a 0.2-ml PCR tube by mouth pipetting. Firststrand cDNA synthesis was performed using a 25-bp oligo(dT)30VN primer for 3′ amplification. PCR products were used to generate second-strand cDNA.After annealing to an index primer,the second-strand cDNA was fragmented into 350-bp pieces by a Bioruptor Sonication System(UCD300,Diagenode, Brussels, Belgium), and the reactions were purified by incubation with Ampure XP beads (Beckman,A63880, Fullerton, California, USA) at room temperature for 5 min. After quality inspection using an Agilent 2100 High Sensitivity DNA Assay Kit(Catolog No.5067-4626,Agilent, Santa Rosa, CA) based on the manufacturer’s instructions,sequencing was performed on an Illumina HiSeq 2000 platform using 150-bp paired-end sequencing via the Smart-seq2 protocol.
Processing of scRNA-seq data
Trimmed reads were aligned to the reference pig genome(genome build: Sscrofa 11.1) using Hisat2 (v2.0.5). The uniquely mapped reads were calculated and partitioned using StringTie (v1.3.5) and Ballgown (v2.16.0) [62]. The transcript counts of each cell were normalized to FPKM.Overall, 760 individual cells were collected for single-cell cDNA amplification, and 464 cells passed the quality control criteria.On average,there were 22 million mapped reads and 7253 detected genes for each cell.
Identification of cell types
The Seurat (v3.0), dplyr (v0.7.0), and umap (v0.2.3.1)packages in R were applied to classify the 464 single cells into major cell types[63,64].Only cell sample with a gene expression number>2000 was considered,and only genes with normalized expression levels greater than one and expressed in at least three cells were retained.Finally,we obtained a total of 22,517 genes across the 464 cells for clustering analysis. Principal component analysis (PCA)of the genes from the 464 cells was conducted using the‘FindVariableFeatures’ function (selection.method =“vst”,nfeatures=2000).Significant principal components(PCs)selected by a JackStraw test with 100 replicates were used to perform the clustering.The first 10 PCs were used to perform uniform manifold approximation and projection (UMAP) based on the ‘RunUMAP’ and ‘FindClusters’ functions. We obtained six cell clusters for TV, LV,and RP.
Identification of cell type-specific expressed genes
Genes that were differentially expressed in each cluster were identified using the ‘FindAllMarkers’ function in Seurat against the normalized gene expression data and were then tested by‘roc’and DESeq2[61].Here,both‘min.pct’and‘thresh.use’values greater than 0.25 were selected as the cut-off for gene selection. SAMtools and BEDTools were used to calculate sequencing depth and reads harboring strand-specific poly(A) tail of each TLV cell,respectively [65,66]. The database for annotation, visualization, and integrated discovery bioinformatics resource(DAVID;a gene functional classification tool)was used for functional annotation and GO analysis [67].
Pseudo-time analysis
The Monocle2 package was used to analyze pseudo-time trajectories to predict developmental processes of TV, LV,and RP cells [68]. We used cell type-specific expressed genes identified by the‘FindConservedMarkers’function in Seurat to sort cells into pseudo-time order.‘DDRTree’was applied to reduce the dimensions, and the visualization functions ‘plot_cell_trajectory’, ‘plot_genes_branched_pseudotime’, and ‘plot_genes_branched_heatmap’ were used to display the branched trajectory, pseudo-time, and heatmap, respectively.
RNA velocity analysis
Read annotations for the Smart-seq2 output data were performed using the velocyto.py command-line tools according to the manual [69]. Genome annotations Sscrofa11.1 and Sscrofa11.1.101 from Ensembl were used to count and sort reads into three categories: ‘spliced’, ‘unspliced’, and‘ambiguous’. The loom file generated was loaded into velocyto.R. Finally, coordinates from the Seurat’s UMAP analysis were used to embed the velocity results.
WGCNA and gene correlation network construction
WGCNAwas performed on the normalized gene expression data measured in FPKM, using unsigned correlation, softthreshold power of six, and minimum module size of 120 members[70].We then generated an independent list of hub genes (eigengene connectivity > 0.9) for each skeletal region. Finally, the co-expression gene network was visualized using VisANT and Cytoscape [71,72]. The Benjamini–Hochberg method was used to correct multiple comparison when calculating the significance of the correlations among modules.
Immunofluorescence staining analysis
A pig embryo at 27 dpf was fixed overnight in 10% neutral formalin-fixed solution at room temperature. Thin 5-μm TV and LV paraffin-embedded sections were collected for immunofluorescence staining. Cell nuclei were stained using DAPI(Catalog No.62247,Life Technologies,Carlsbad,CA).Primary antibodies used were: MATN1 (Catalog No.orb94279, Biorbyt, Cambridgeshire, UK), HMGB2 (Catalog No. ab67282, Abcam, Cambridgeshire, UK), COL1A1(Catalog No. ab34710, Abcam), CD34 (Catalog No.orb348961, Biorbyt), CD93 (Catalog No. ab198854, Abcam), and CD248 (Catalog No. sc-377221, Santa Cruz Biotechnology, Delaware, CA). The secondary antibody used was anti-rabbit IgG (Catalog No. ZDR-5003, ZSGBBIO, Beijing,China).
Image analysis and data processing
Images of the paraffin sections were collected by digitizing the images using a Leica Aperio Versa 200 slide scanner.All images were processed using ImageScope.
Ethical statement
Pig embryonic, fetal sample collection, and single-cell transcriptome study were carried out under the approval of the Kunming Institute of Zoology, Chinese Academy of Sciences, China (SMKX-20191213-01). All experiments were done following the International Review Board, Institutional Animal Care,and Use Committee guidelines.
Data availability
scRNA-seq data generaged in this study have been deposited in the Genome Sequence Archive [73] at the National Genomics Data Center, Beijing Institute of Genomics,Chinese Academy of Sciences/China National Center for Bioinformation (GSA: CRA002562), and are publicly accessible at https://ngdc.cncb.ac.cn/gsa.
CRediT author statement
Jianbo Li:Data curation,Writing-original draft,Writingreview & editing.Ligang Wang:Data curation, Writing -original draft.Dawei Yu:Conceptualization,Methodology,Visualization.Junfeng Hao:Conceptualization, Methodology, Visualization.Longchao Zhang:Conceptualization,Methodology.Adeniyi C.Adeola:Writing-review&editing.Bingyu Mao:Writing - review & editing.Yun Gao:Investigation.Shifang Wu:Investigation.Chunling Zhu:Investigation.Yongqing Zhang:Writing - review &editing.Jilong Ren:Investigation.Changgai Mu:Investigation.David M. Irwin:Writing - review & editing.Lixian Wang:Data curation,Writing-original draft.Tang Hai:Data curation, Writing - original draft.Haibing Xie:Data curation,Writing-review&editing.Yaping Zhang:Data curation, Writing - original draft, Writing - review &editing. All authors have read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by the Strategic Pioneer Program of the Chinese Academy of Sciences (Grant No.XDA24010107), the Ministry of Agriculture of China(Grant No. 2016ZX08009003-006), the China Agriculture Research System (Grant No. CARS-35), and the Agricultural Science and Technology Innovation Project,China(Grant No.ASTIP-IAS02).This work was supported by the Animal Branch of the Germplasm Bank of Wild Species,Chinese Academy of Sciences (the Large Research Infrastructure Funding).
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2021.09.008.
ORCID
0000-0002-6431-6421 (Jianbo Li)
0000-0002-4376-8373 (Ligang Wang)
0000-0002-4504-7254 (Dawei Yu)
0000-0002-8209-5247 (Junfeng Hao)
0000-0002-4520-3165 (Longchao Zhang)
0000-0001-5171-7878 (Adeniyi C. Adeola)
0000-0002-7993-3158 (Bingyu Mao)
0000-0002-5148-7598 (Yun Gao)
0000-0001-9343-6521 (Shifang Wu)
0000-0001-7356-3419 (Chunling Zhu)
0000-0003-0581-4882 (Yongqing Zhang)
0000-0002-2400-1951 (Jilong Ren)
0000-0002-6256-9118(Changgai Mu)
0000-0001-6131-4933 (David M. Irwin)
0000-0001-8943-4697 (Lixian Wang)
0000-0002-3192-0753 (Tang Hai)
0000-0003-4977-8270 (Haibing Xie)
0000-0002-5401-1114 (Yaping Zhang)
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Genomics, Proteomics & Bioinformatics
- Call for Papers Special Issue on “Artificial Intelligence in Omics”
- Call for Papers Special Issue on “RNA Modifications and Epitranscriptomics”
- Advanced Single-cell Omics Technologies and Informatics Tools for Genomics, Proteomics, and Bioinformatics Analysis
- A Single-cell Transcriptome Atlas of Cashmere Goat Hair Follicle Morphogenesis
- Integration of Droplet Microfluidic Tools for Single-cell Functional Metagenomics: An Engineering Head Start