Integration of Droplet Microfluidic Tools for Single-cell Functional Metagenomics: An Engineering Head Start
2021-02-24DavidConchousoAmaniAlMaabadiHayedehBehzadMohammedAlarawiMasahitoHosokawaYoheiNishikawaHarukoTakeyamaKatsuhikoMinetaTakashiGojobori
David Conchouso, Amani Al-Ma’abadi, Hayedeh Behzad, Mohammed Alarawi, Masahito Hosokawa, Yohei Nishikawa, Haruko Takeyama,7,Katsuhiko Mineta, Takashi Gojobori,
1Department of Industrial and Mechanical Engineering, Universidad de las Américas Puebla, Puebla 72810, Mexico
2Computational Bioscience Research Center, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
3Biological and Environmental Sciences and Engineering Division,King Abdullah University of Science and Technology,Thuwal 23955-6900, Saudi Arabia
4Research Organization for Nano & Life Innovation, Waseda University, Tokyo 162-0041, Japan
5Department of Life Science and Medical Bioscience, Waseda University, Tokyo 162-8480, Japan
6Institute for Advanced Research of Biosystem Dynamics, Waseda Research Institute for Science and Engineering, Waseda University,Tokyo 169-8555, Japan
7Computational Bio Big-Data Open Innovation Laboratory, AIST-Waseda University, Tokyo 169–0072, Japan
8Computer, Electrical, and Mathematical Sciences and Engineering Division, King Abdullah University of Science and Technology,Thuwal 23955-6900, Saudi Arabia
Abstract Droplet microfluidic techniques have shown promising outcome to study single cells at high throughput.However, their adoption in laboratories studying “-omics” sciences is still irrelevant due to the complex and multidisciplinary nature of the field. To facilitate their use, here we provide engineering details and organized protocols for integrating three droplet-based microfluidic technologies into the metagenomic pipeline to enable functional screening of bioproducts at high throughput. First, a device encapsulating single cells in droplets at a rate of~250 Hz is described considering droplet size and cell growth. Then, we expand on previously reported fluorescence-activated droplet sorting systems to integrate the use of 4 independent fluorescence-exciting lasers (i.e., 405, 488, 561, and 637 nm) in a single platform to make it compatible with different fluorescence-emitting biosensors.For this sorter,both hardware and software are provided and optimized for effortlessly sorting droplets at 60 Hz.Then,a passive droplet merger is also integrated into our pipeline to enable adding new reagents to already-made droplets at a rate of 200 Hz.Finally,we provide an optimized recipe for manufacturing these chips using silicon dry-etching tools. Because of the overall integration and the technical details presented here,our approach allows biologists to quickly use microfluidic technologies and achieve both single-cell resolution and high-throughput capability (>50,000 cells/day)for mining and bioprospecting metagenomic data.
KEYWORDS Droplet microfluidics;Single cell;Metagenomics;Droplet sorter;Biotechnology
Introduction
Droplet microfluidic technologies are revolutionary tools enabling chemical and biological discoveries [1–5]. In biology, droplet microfluidics has an extraordinary impact on developing novel assays performed at the granularity of a single cell and high throughput [6–12]. For example, it is now possible to carry out millions of single-cell studies in microfluidic droplets acting as microreactors of only~ 1–1000 pl each [13]. In these droplets, one can encapsulate single cells [6,11], insert controlled amounts of reagents [7,12–15], add unique barcodes to each droplet[7,8,16,17],and deliver biosensors to detect cell characteristics at unprecedented precision and high-throughput capability[6,7,11,18].
Other characteristics that are key for the integration of these technologies into biological research include accurate detection of biological products[5,11,15,18–20],rapid heat transfer and cycling [21], low-cost [22,23], as well as simple integration with automation, data processing tools[6,8,18,24],and portable electronic devices[25].
These features create new opportunities for developing ambitious protocols to mine and discover novel genes [11]and entire genomes of environmental microorganisms[26,27].The potential of droplet microfluidics is massive,as the pool of environmental microbes contains trillions of unique life forms [28,29], each containing fundamental pieces of information in their genes that are the key to understanding the mechanisms of evolution and discovering new biochemical products. These protocols can also study cell-to-cell heterogeneity between cells belonging to the same microbial population [27,30] or the same tissue or organ[8].Consequently,droplet microfluidics can open the doors to numerous unexplored areas.
Traditional methods cannot practically handle single-cell studies,as 99%of the microorganisms cannot be cultured in the laboratory[29]and their throughput is low compared to the number of single-cell experiments needed. Some techniques, such as 16S rRNA gene sequencing, can obtain phylogenetic relations of uncultured microorganisms [31]but cannot mine genes for bioprospecting. Other metagenomic techniques enable gene mining from environmental microbiomes by cloning exogenous DNA in surrogate host cells to perform sequence-based and function-driven analyses in a well-controlled host system [29]. Nevertheless,these methods still require many manual operations,making them impractical.
Droplet microfluidics is the only technology capable of providing the high-throughput analysis required from sample collection to sequencing.Moreover,this technology also offers seamless integration with automation[20,32,33]and digital processing tools that are vital for analyzing the Big Data produced by the gigantic pool of living microorganisms and next-generation sequencing(NGS)techniques[29,34,35].
Despite the evidence of successful microfluidic protocols for studying single cells at unprecedented throughput and resolution [6–8,11,12], only a few laboratories working on“Lab on a Chip”use them.Some of the reasons behind this poor implementation are:1)the limited access of biologists to cleanroom facilities and the equipment needed to make microfluidic chips and conduct experiments;2)the complex and multidisciplinary nature of microfluidics (e.g., microbiology,biosensing,NGS,bioinformatics,fluid mechanics,electronics, controls, and nanofabrication); 3) the lack of standardized protocols and methodologies; and 4) the significant budget required to start a single-cell microfluidics facility.This study will focus on solving points 2)and 3)by providing critical information and engineering insights to facilitate biologists using droplet microfluidics.
Our group studies the metagenomic diversity of the Red Sea because of its unique conditions such as high salinity and high temperature [28,30,34–37]. During this process,we assembled the set of droplet microfluidic tools, here described, to perform single-cell studies directly from metagenomic libraries of the Red Sea. Including the automatic screening of these libraries based on phenotypic responses (i.e., functional metagenomics), we selectively carried out NGS and identified new species and biological products.This study presents the integration of these tools and provides technical engineering details to implement these tools in biological laboratories. The chosen methods include the main functions needed for single-cell metagenomic studies (Figure 1): 1) single-cell encapsulation in microfluidic droplets using droplet generators with two aqueous inlets(Figure 1A)or one aqueous inlet(Figure 1B)[6,11], 2) fluorescence-based droplet sorting (Figure 1C)[6], and 3) passive droplet merging, which enables the addition of precise amounts of reagents to the droplets(Figure 1D) [12].

Figure 1 Droplet microfluidic techniques reviewed and integrated in this studyA.Droplet generation device for single-cell encapsulation with two inlets for introducing two different sample fluids.B.Droplet generation chip with only one inlet for the sample fluid. Both droplet generators can produce droplets at rates of 250–350 droplets/s and effectively encapsulate single cells at concentration of 0.1 cells/droplet. C. Automatic droplet sorting chip based on fluorescence. The high voltage signal applied to the terminals creates a dielectrophoretic effect, resulting in droplet sorting. The electrodes conducting the signal are colored in red and black. This picture also shows the interrogation window,where fluorescence inspection is taking place by shining each droplet with a laser and capturing the emitted light with a PMT sensor.D.Passive droplet merging device capable of merging droplets at 200 droplets/s.Note.All the channels in the insets colored in blue are filled with the oil phase, whereas channels colored in orange carry the sample fluids’ aqueous phases. The designs of these devices were adapted from [6,12]. Panels A–C were adapted by permission from Mazutis and colleagues [6]. PMT, photomultiplier tube; HVA, high voltage amplifier.
First, a guide for manufacturing these devices is provided. This guide includes recipes for producing longlasting molds on silicon using dry etching techniques.Then,single-cell encapsulation is discussed to identify critical problems arising from this process, and a guidance is provided to solve them.Later, we present technical details for building a fully automated droplet sorter using fluorescence signals, electronic controls, lasers, and optics. We also provide the program developed for this procedure,insights about using the electronic instruments, and a summary of the suggested lenses and mirrors for different fluorescenceemitting biosensors working at distinctive excitation and emission wavelengths. Additionally, a passive droplet merger is described, and details of its operation are examined. Finally, we provide a discussion on the overall integration of these methods toward functional metagenomic studies.
This study is an engineering head start for any laboratory willing to incorporate droplet microfluidics into their research, because it gives a step-by-step guide to implement and integrate droplet microfluidic techniques into the metagenomic pipeline.
Method
Materials, equipment, and consumables used in this study
Table 1shows a list of the materials and equipment used in this study, including reagents, optical components, devicefabrication materials, and microfluidic and electronic equipment. We also report on model numbers, names, and locations of the vendors to quickly obtain the exact products.A picture of the facility designed to integrate the three microfluidic technologies, here described, is shown in Figure S1.
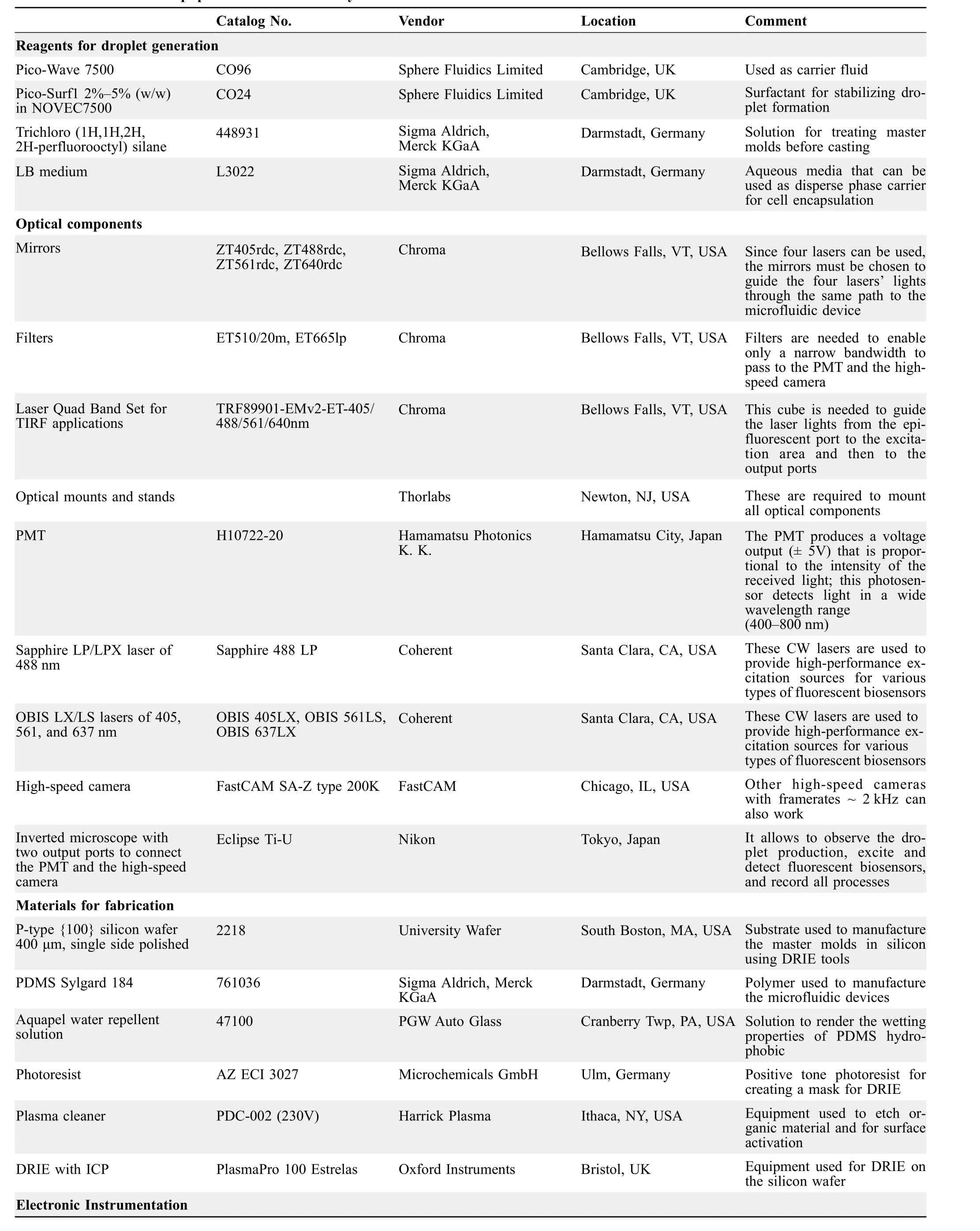
Table 1 Materials and equipment used in this study

Table 2 Process parameters for DRIE etching the master molds on silicon
(Continued)
The MFCS series pressure controllers from Fluigent are used to drive the fluids through the microfluidic chips.These controllers have a pressure range from 0 to 700 kPa and were managed through the company’s proprietary software MAESFLO. The flow rates were measured using flow-rate sensors from the same company. Although these sensors are usually calibrated for water, it is possible to calibrate them to work with other fluids using a proportionality parameter.
Fabrication method for manufacturing silicon master molds for soft lithography
Various manufacturing techniques for microfluidic devices were developed using techniques borrowed from the microelectro-mechanical system (MEMS) industry. Examples include lasers and conventional milling machines[32,33,38,39], hot embossing [40], injection molding [41],digital light-projecting [42], 3D printing [43,44], capillary assembly [45], poly(ethylene)-glycol-diacrylate (PEGDA)casting [46],and paper-based patterning amongst others.Soft lithography[6,47,48]is the most popular technique for fabricating microfluidic chips. This process involves creating high-quality master molds for casting polydimethylsiloxane (PDMS) replicas. Although soft lithography was initially designed as a rapid prototyping technique for testing microfluidic designs,several protocols and commercially available products consider using PDMS chips in single-cell studies because of the following advantages:PDMS devices are affordable,the replicas inherit the high resolution of master molds, PDMS is biocompatible, and there are several available surface-crafting techniques to render the wetting properties[49–52].
A disadvantage of using PDMS is the low reusability of the devices in biological applications. One cannot use the same chip twice due to cross-contamination between experiments.Therefore,many PDMS chips are required daily,and consequently,the master molds must last for numerous casting processes. In this situation, the master mold’s lifetime is critical. Here, we describe the most common challenges associated with the fabrication of these molds by two methods, SU-8 lithography and bulk micromachining in silicon.
Fabrication challenges and benefits of SU-8 master molds
SU-8 lithography is an excellent technique for making master molds for soft lithography,because SU-8 photoresist can be deposited at different thicknesses(e.g.,0.5–200 μm)and can also be patterned at high-aspect ratios (usually> 10:1).This method also allows for creating multi-height structures with a smooth surface, making it a versatile technology for creating high-quality masters[6,53].
Although SU-8 presents excellent advantages for rapid prototyping and low-cost manufacturing of masters, depositing thick SU-8 layers (> 50 μm) can become challenging for someone unfamiliar with photolithography due to the high viscosity of the photoresist. An inexperienced person may face problems with thick SU-8 layers, such as trapped bubbles,nonuniform spin-coating,poor adhesion to the wafer, incorrect UV exposure times, internal stresses[54,55], and poor mask contact resulting in low-feature resolution due to diffraction[56].
SU-8 masters can be used for many PDMS casting procedures, demonstrating excellent lifetime usage. However,sometimes these molds can show signs of wear, and the structures can be peeled off after some casting cycles due to any of the fabrication challenges mentioned above.
Fabrication of silicon-based master molds using dry etching tools
Another type of master molds,with a long-lasting lifetime,is made by bulk micromachining directly on a silicon wafer using deep reactive ion etching (DRIE) and cleanroom facilities [57,58]. Although this process requires specialized equipment and not all the laboratories have access to it,the fabrication of this type of master molds becomes less variable and more robust than SU-8.
These molds can be used indefinitely to make PDMS chips, showing exceptional lifetime usage as features are made in bulk silicon.The DRIE process is not trivial,but we provide a recipe for Oxford Instruments’ DRIE tool with inductively coupled plasma (ICP) as a starting point for anybody who wants to manufacture these devices for the first time.
The process begins with a new p-type {100} silicon wafer,in which an ECI-3027 photoresist mask is deposited and patterned using standard lithography. We selectively etch the silicon and create high-aspect-ratio features directly onto the silicon substrate(Figure 2).The process details are described below.

Figure 2 Fabrication of silicon-based master molds and PDMS chipsA.SEM image of the master mold for a droplet generator device.The channel features are well defined with smooth surfaces and straight walls.B.Casting of PDMS over the master mold.We prepared a mixture of the polymeric base and the curing agent(10:1)as suggested by the Sylgard®184 Silicone-based Elastomeric Kit. This mixture was degassed in a vacuum chamber and then poured on top of the silicon master. Once the PDMS mixture is fully crosslinked, the inlets/outlets are punched, and the resulting PDMS replica is bonded to a glass slide using O2 plasma treatment. C. The inset presents an example of a finalized chip. SEM, scanning electron microscopy; PDMS, polydimethylsiloxane.
Mask printing
This process was performed using a μPG-101 from Heidelberg Instruments.The mask designs were modified from previous studies[6,12],and they were printed in their light field configuration for positive photoresist(i.e.,opposite to SU-8 masking). A copy of the mask files for photolithography is provided as Datasets S1–S3.
Wafer pretreatment(optional)
The wafer was pretreated to improve the adhesion of the photoresist by removing the existing natural oxide layer.This treatment was achieved by submerging the wafer in a 20-s buffered oxide etch (BOE) bath, rinsing with deionized (DI) water for 10 min, and drying on a hot plate. Warning, BOE contains hydrofluoric (HF) acid,and improper handling may result in serious injuries or death.A second possible pretreatment is to deposit a thin film of the hexamethyldisilizane (HMDS) adhesion promoter; however, this process requires specialized equipment [59].
Standard photolithography
A 4-μm layer of ECI-3027 positive photoresist was spun,prebaked, exposed, and developed to act as a mask to etch the silicon wafer selectively. Since this is a standard procedure, the resulting lithography was uniform and welldefined.
DRIE
This process consists of a series of etching and deposition(i.e., passivation) cycles that alternate continuously to produce high-aspect structures on silicon. A summary of the process parameters is shown inTable 2. The initial conditions for the process were set to achieve a high vacuum(e.g., 7.5 × 10−9Torr) and reach a stable temperature of−20 ºC. The number of cycle repetitions depends on the feature density in our wafer. If the feature density is low,there is a larger surface area to etch away,and therefore,the etching rate is slow (e.g.,~0.328 μm/cycle for the droplet generator chip). Conversely, if the feature density is high,there is less silicon to remove, and the etching rate is fast(~ 0.4 μm/cycle for the droplet sorter chip). In any case, a feature with a height of 50 μm takes from~120–150 cycles.Figure 2A shows a scanning electron microscopy (SEM)image of the silicon mold.
Photoresist strip
The remaining ECI-3027 photoresist was then stripped away using acetone.
Oxygen plasma clean In the case of any remaining photoresist left in the silicon wafer,30-s O2plasma clean is recommended.This process also helps to activate the surface in preparation for the next step.
Pretreatment for PDMS casting
The masters were treated with trichloro (1H,1H,2H,2Hperfluorooctyl) silane to facilitate demolding. A drop of 50–100 μl was poured into a container and placed next to the master in a closed chamber.A vacuum was then applied for 4 h to evaporate and deposit the silane on the master’s substrate. Caution, silane is toxic, and this step must be performed under a fume hood.
PDMS casting (Figure 2B)and bonding
After the master was completed, one can follow the standard soft lithography procedures [6,47,48].
Fabricated devices are shown in Figure 2C. To store these chips, we recommend covering all surfaces with frosted Scotch tape until their first use to avoid capturing dust and debris on the chip’s surface.Finally,the channels’walls were rendered to favor the wetting properties of the carrier fluid. Since fluorocarbon (FC) oils such as FC-40 and HFE-7500 are commonly used in single-cell experiments,one must render the PDMS channels hydrophobic.A couple of methods to achieve this surface modification include flushing the devices with Aquapel water-repellent solution(30 s)[6,7]or with a 1%trichloro(1H,1H,2H,2Hperfluorooctyl) silane solution in HFE-7500 oil (v/v)[60,61]. In both cases, the devices must be rinsed with the oil and blow-dried immediately after treatment [11,12,62].This rinsing step is of utmost importance for Aquapel treatment because this solution can quickly react with oxygen and form particles that clog the devices.
Single-cell encapsulation in droplet microfluidics
Single-cell encapsulation is the basis of this research. Millions of cells can be individually analyzed in a pico-liter(pl)droplet that acts like a micro-Eppendorf tube where only the appropriate reagents and a single cell are present. Two critical factors are relevant for encapsulating only one cell per droplet:generation of droplets with stable and uniform size and a constant cell concentration in the sample fluid[6,11].Here,we describe some of the possible solutions to address these challenges.
Microfluidic droplet generation results from the interaction between two immiscible fluids forced to flow together inside a microchannel.Droplet formation then occurs when the interfacial tension and the fluid’s viscous force are balanced [32]. Although there are different channel configurations for droplet generation [5], flow-focusing generators are frequently preferred for single-cell encapsulation [6–8,11,63]. In this configuration, the droplet size heavily depends on the flow rate ratio of two liquids.The higher the oil-water flow rate ratio is, the smaller the produced droplets [64]. The droplet’s volume is proportional to the cubic of the characteristic length;therefore,one must carefully choose a droplet size and keep it constant throughout the experiments. For example, a droplet of 100 μm in diameter has a volume of 524 pl,whereas a 40-μm droplet has only 33.5 pl. Therefore, a slight difference in droplet size can directly affect the encapsulation efficiency.
Additionally, if droplets have different sizes, we may compromise the droplets’operational handling in the other two processes (i.e., droplet sorting or merging). Therefore,the size of the droplets must be carefully designed. In the case of our experiments, the droplet size was 45 μm in diameter.
The stability of the droplets is addressed by modifying the surface tension between the liquids.Usually,this can be achieved using surfactants such as 2%–5% (w/w) Pico-Surf1 [11] (Sphere Fluidics Limited, Cambridge, UK) or 2%(w/w)deprotonated Krytox [7] surfactant in FC oils.
Cell concentration can be measured optically using a bright-field microscope and a hemocytometer or electronically with a cell counter. The cell concentration must be 0.1 cells/droplet to avoid encapsulating more than one cell per droplet [11,65]. Working with living cells in nutrientrich solutions [66,67], such asE.coli-based metagenomic libraries in lysogeny broth (LB) medium, brings other complications to cell encapsulation efficiency.Cell division can occur in 20–60 min [68], and therefore, the cell concentration may not remain constant at 0.1 cells/droplet during the encapsulation process. Cell division can be delayed by lowering the temperature of the tubing in an ice bath. If cell concentration varies due to gravity, adding OptiPrep density gradient medium from Sigma-Aldrich can reduce this problem.
Fluorescence-activated droplet sorting
One of the biggest challenges of working simultaneously with millions of droplets (i.e., experiments) is to quickly screen positive droplets from negative ones. This task requires automation and electronics to properly handle the throughput needed[69–71].Here,we describe six essential elements to sort the droplets selectively: detection signal,control system, actuation mechanism, connection diagram and flow of information, optics and fluorescent excitation using lasers, and software development[71].
Detection signal
In metagenomics, fluorescent biosensors are commonly used to detect a phenotypic response in cells. These biosensors can be chosen based on particular enzymatic functions;thus,they can help us narrow down the specific cells of interest with a high probability of obtaining novel findings.In this regard,fluorescent biosensors are vital elements that enable the functional detection of targeted droplets.
Control system
A control system is a decision-making unit enabling time synchronization between detecting a positive droplet and switching on the actuation mechanism [71]. The system’s response time is a critical design parameter to achieve efficient sorting.The overall response time must consider the reaction time of the electronic instruments, and the time required by the actuation mechanism. Those two times should then be synchronized with the speed of the moving droplets.
Actuation mechanism
Several actuation mechanisms can be used to manipulate droplets in microfluidic channels. These mechanisms include magnetic field manipulation,pneumatic control, dielectrophoretic action, acoustic wave interactions, and thermally actuated mechanisms [71]. Among these techniques, dielectrophoretic action was chosen in this study,because it offers the fastest actuation time (e.g., sorting up to 30 kHz [69]) and is the best candidate for single-cell screening at high throughput [6,69,72]. Dielectrophoretic sorting consists of selectively manipulating the droplets by activating a high-voltage alternating current (AC) electric field in a localized area of a microfluidic channel.This field induces a dipole moment to slightly push a positive droplet to a different channel using a“Y-junction” [6,69,72].
Connection diagram and flow of information
We show the equipment and connections to successfully implement the dielectrophoretic droplet sorter in this subsection.The design of the sorter was obtained from Mazutis and colleagues[6].However,we focus on expanding some technical details because biologists are generally unfamiliar with these electronic components.Figure 3shows the flow of information from one component to the other. The process starts by detecting a fluorescence signal and ends when the high-voltage AC signal is triggered at the microfluidic chip,sorting the positive droplets out.The processing time for executing these electronic commands across the different instruments was measured to be 360 μs. The effective sorting rate achieved with this system was limited by the speed of the passing droplets but not by the electronic instrumentation.
Optics and fluorescent excitation using lasers
The implementation of four laser sources (i.e., 405, 488,561, and 637 nm) for exciting different fluorescence-emitting light biosensors using the same setup is presented inFigure 4. By selecting mirrors that reflect specific wavelengths and transmit the others(Table 3),one can combine the light path of four or more lasers,as shown in Figure 4A.Alternatively,these lasers can be mounted on an OBIS LX/LS Laser box,and then the lasers’light can be guided to an inverted microscope by optical fibers.
Once excitation light arrives at the inverted microscope,it hits a filter box and then goes to the sorting chip through the objective lens. The emitted light then bounces back across the filter box and arrives at the high-speed camera and the photomultiplier tube (PMT) ports at a ratio of 90%:10%(Figure 4B).The filter box is at the center of the microscope and selectively filters and directs the excitation and emitting lights to the right locations.The filter box is a set of components composed of an excitation filter, an emission filter, and a dichroic mirror. Each of these components must be chosen depending on the excitation and emitting wavelengths of the biosensor. Fortunately, there are filter box sets that are compatible with two or more laser wavelengths. For example, the Laser Quad Band Set from Chroma (Catalog No. TRF89901-EMv2-ET-405/488/561/640nm) can be used with the four proposed laser wavelengths.
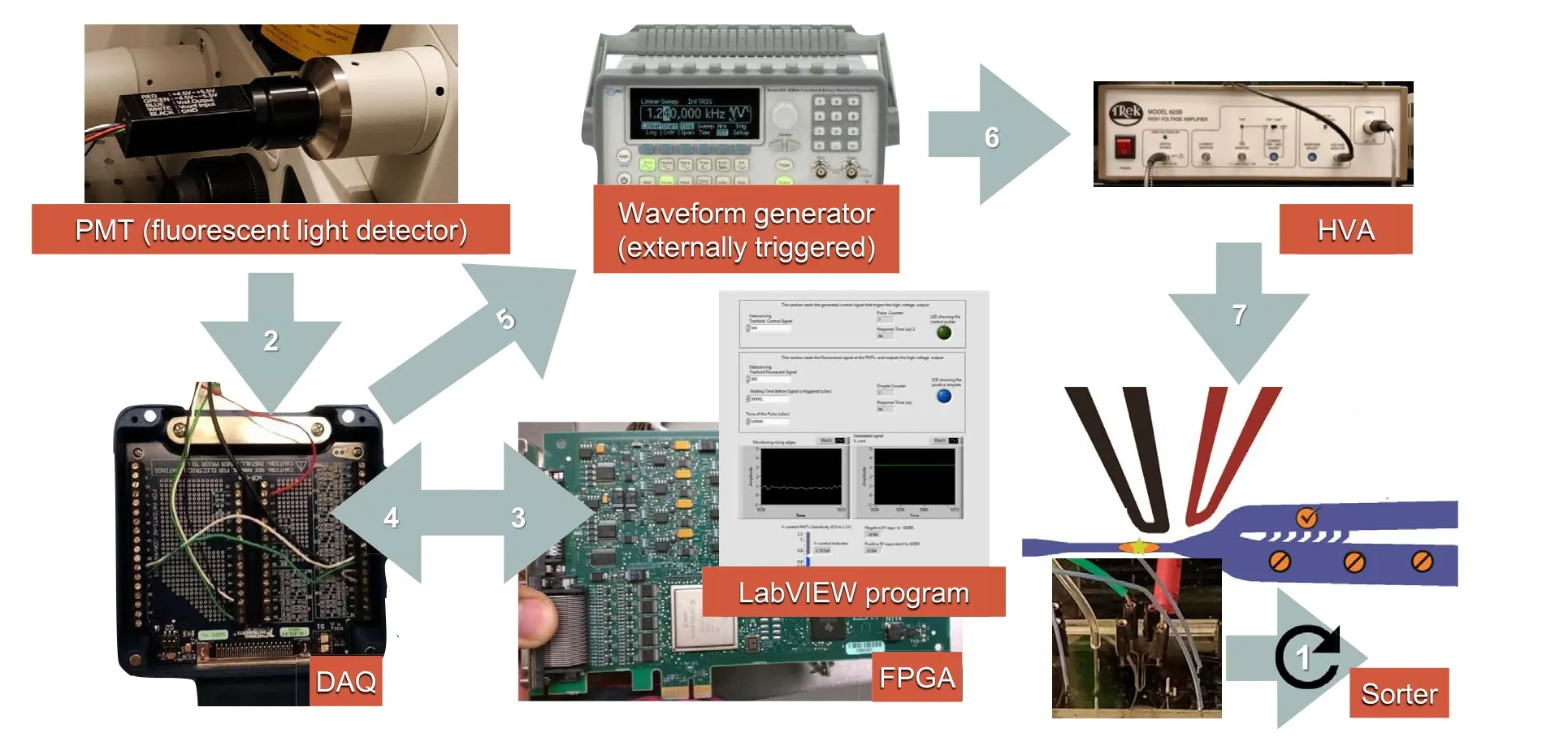
Figure 3 Flow of information through the automatic droplet sorting chipWhen the PMT sensor detects fluorescent light,it sends a“true”signal to the FPGA board through the DAQ board.The FPGA logic board then decides to send back a triggering signal to the waveform generator (externally triggerred) also through the DAQ board. On this command, the waveform generator creates a train of 600 pulses at a frequency of 30 kHz, with an amplitude of 500 mVpp and a DC offset of 250 mV. Simultaneously, the output of the waveform generator is amplified at a ratio of 1:1000 by the HVA and sent to the microfluidic chip terminals.The reaction time for the electric signals was measured to be 360 μs. FPGA, field programable gate array; DAQ, data acquisition; DC, direct current.
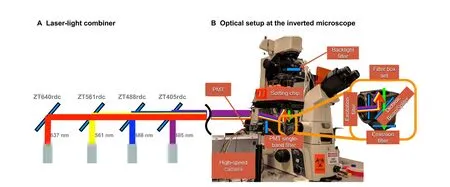
Figure 4 Lasers’ path and optical setup for fluorescence detectionA. Laser-light combiner. Dichroic mirrors are used to align the laser beams in a single path guided to the epi-fluorescent illumination port of the microscope. The mirrors are selected to pass the light wavelengths to their left and reflect the wavelength of the corresponding laser. Using this configuration,one can excite the droplets with one or more laser sources.B.Optical setup and light’s path through the inverted microscope.The excitation light travels from the input port to the sample chip,and the emitting light goes from the chip to the high-speed camera and the PMT ports.A key element reflecting and filtering the light is the filter box placed at the inverted microscope center.
Connecting a high-speed camera and a PMT sensor at the same time is challenging.On the one hand,the high-speed camera requires an intense illumination backlight to record high-speed video. On the other hand, the PMT sensor is extremely sensitive, and it gets quickly saturated even with ambient light. For this reason, we use a beam splitter of a 90%:10%ratio to send most of the light to the camera port.Additionally,we installed a backlight filter and a PMTsingleband filter to allow only a narrow wavelength band to pass through each sensing device.These two filters are required to have independent wavelength bands, and their selection depends on the fluorescent biosensor of interest.For example,a biosensor emitting 517 nm light can use an ET510/20m as a PMT filter and an ET665lp as a backlight filter.
Since the four laser sources are installed on the same platform,they can be used simultaneously or independently to excite the passing droplets. However, the sorting device has only a single “Y-junction”, and therefore, it can only sort droplets in two different groups:fluorescence-emitting droplets and non-fluorescence-emitting droplets. If more than two groups are required,one must repeat the process as many times as different wavelengths exist.
Software development
The use of field programmable gate arrays (FPGAs) has enabled the design of mid-to-high-complexity systems in a programable and manageable way [73].Since droplet sorting requires fast response times (μs range), we decided to implement the sorting controls on LabVIEW FPGA hardware(Figure 3).The code running on this device has a clock cycle of only 25 ns, thus allowing our entire program to execute in only 66 μs.This program comprises three subroutines and a user interface as depicted in Figures S2–S5.Figure S2 shows the subroutine for configuring and initializing the PMT, and Figure S3 illustrates the subroutine for reading the PMT sensor and sending a triggering signal to the function generator. Likewise, Figure S4 presents a subroutine for simulating the detection of a positive droplet,and finally,Figure S5 shows the overall user interface.
The connection between the microfluidic system and the FPGA hardware was achieved using a data acquisition(DAQ) board (Figure 3).Table 4describes all the wiring required to run the FPGA program and implement the sorting system.
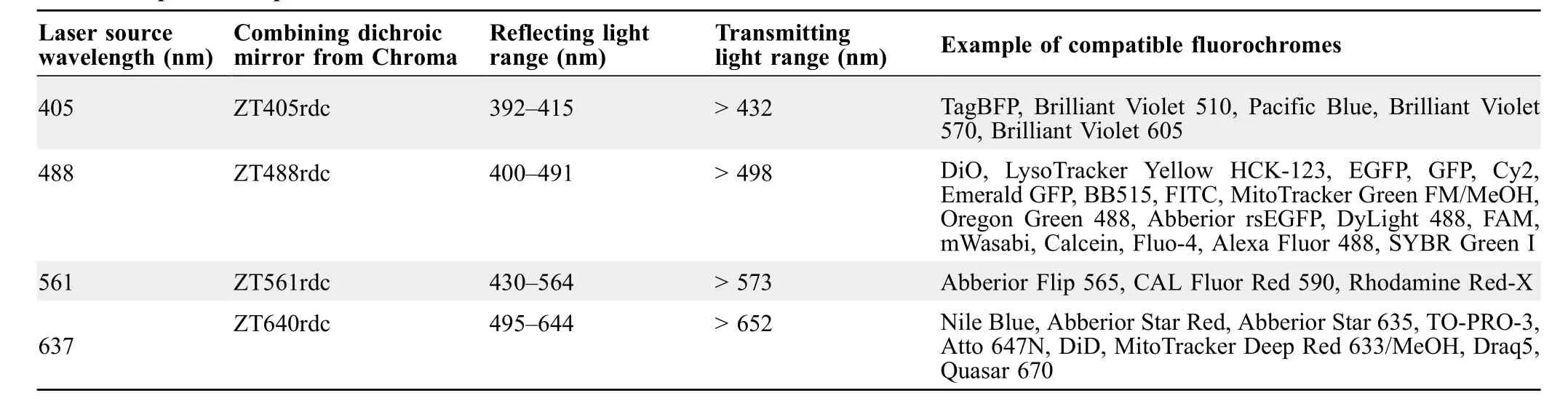
Table 3 Optical set up
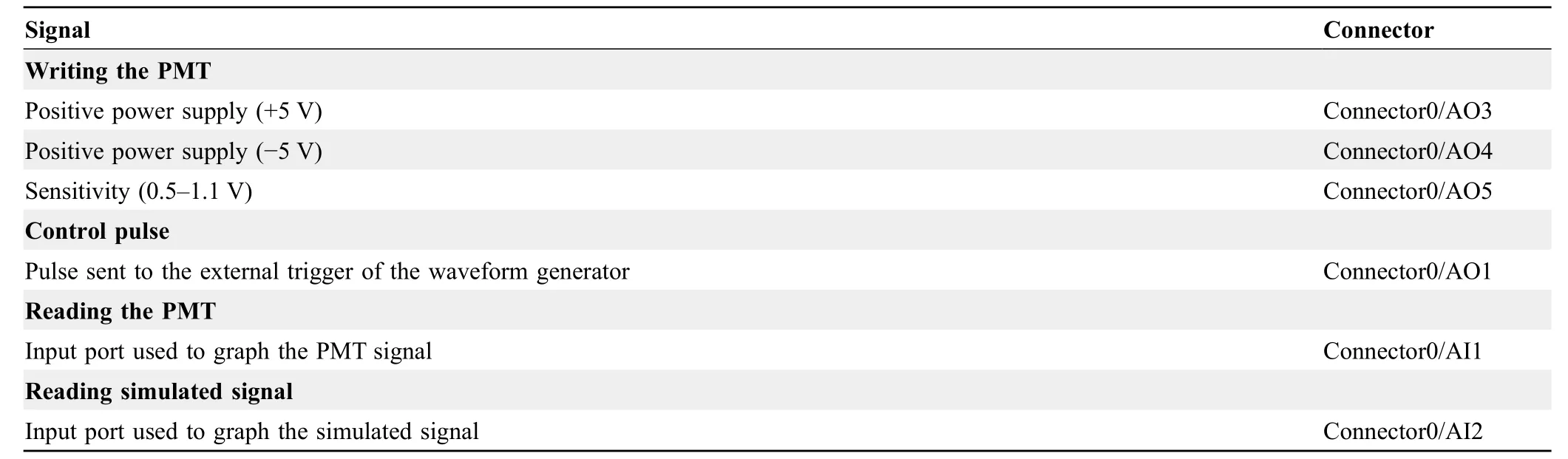
Table 4 Electrical connections required to run the FPGA program
The system also requires a high voltage amplifier(HVA)to produce a large electric field capable of deviating a positive droplet to a different output utilizing dielectrophoresis.
One of the most crucial factors for successfully implementing this sorting device is to measure the time it takes for a droplet to go from the laser interrogation window to the beginning of the splitting channels (Figure 1C). This time is measured with a high-speed camera, and then it is used to synchronize the controls with the droplets’ flow effectively. A useful tool to evaluate droplets’ speed in a channel from a high-speed video was reported by Basu and colleagues [24].
Passive droplet merger for the addition of other reagents
The third droplet microfluidic function,reviewed here,is a passive droplet merger developed by Hosokawa and colleagues [12]. This function enables the addition of precise amounts of reagents to each droplet for further processing.For example,droplet merging has been used to add multiple displacement amplification (MDA) reagents to cell lysate droplets to amplify single-cell genomes [12]. Likewise,others have merged droplets with barcoding DNA sequences[66]to tag and then identify each experiment.In general,droplet merging is a useful tool to enable multiplestep reactions in droplet microfluidics at high throughput.
There are active[7,65,74–77]and passive[12,14,78–80]droplet merging devices. Regardless of the merging approach, 1:1 droplet synchronization is one of the most challenging aspects. Our group has published the experimental details of the device described in this section [12].When this system runs efficiently, it can yield a droplet merging success rate of 95% at a rate as high as 12,000 droplets/min. In this merging device, partially stabilized droplets are merged with newly formed droplets of a particular reagent (e.g., lysis buffer, MDA reagents, and barcoding).Once the droplets are merged at the zigzag channel,an additional surfactant is added to stabilize the newly formed droplets.
Droplet merging often implies reinjecting already-made droplets into a merging device.If one is not careful with this reinjection process,droplets can be poorly packed together and unevenly spaced, leading to synchronization problems when merging in a 1:1 ratio.
For a successful reinjection,the droplet suspension must settle first until most of the droplets aggregate at the top of the container. Then these closely packed droplets can be carefully retrieved using a piece of tubing and a syringe pump with a constant withdrawing rate (e.g., 10 μl/min).Once the tubing is loaded with the droplets,one can detach the tubing with the droplet suspension from the syringe pump and connect it directly to the merging chip.
Results and discussion
In this section,we discuss the overall integration of the three microfluidic techniques presented here and show a practical way for achieving the high-throughput requirements of single-cell functional metagenomics studies.
In functional metagenomics [81,82], characterization of metagenomic fragments is essential for finding applicable values as genetic resources. In practice, functional metagenomics deals with several experimental processes, such as extraction of microbial DNA from environmental samples, construction of an expression library with the microbial genetic material,and a screening method for detecting the desired phenotypic responses. The first two processes are obtained in large quantities(e.g.,millions of samples at a time), and they are discussed elsewhere [83]. However,the third process is more challenging because it involves searching for different phenotypes in each cell in a library of millions. Therefore, traditional techniques in plates cannot practically handle the number of experiments required.The required screening throughput can only be achieved using droplet-based microfluidic methods. For these reasons, in this study, we decided to integrate the three droplet microfluidic techniques to enable a practical screening of functional metagenomic libraries [6,12] in a single platform.This integration can overcome the obstacles that were not adequately addressed previously.
For example,the protocol by Mazutis et al.[6]lacks the capability of adding precise amounts of regents to alreadyformed droplets as these droplets are closed micro-reaction chambers with no extra inputs. Similarly, the protocol by Hosokawa et al. [12] massively overcomes this limitation with a droplet merger,but unfortunately,their throughput is tremendously hindered by manually sorting the droplets with a pipette.
Our combinatory approach of the three droplet microfluidic tools is described inFigure 5and in Movie S1.The output of the first technology is the encapsulation of millions of single cells in microdroplets (Figure 5A). This process is carried out at a cell concentration of 0.1 cells/droplet,and our device generates droplets at 250–350 droplets/s. Therefore, a hundred thousand cells can be encapsulated in approximately 1 h.
With the second technology, one can automatically sort out the droplets that show positive fluorescence (i.e.,functional metagenomic information) from the negative ones (Figure 5B). Our sorting system offers a quick electrical response from fluorescence detection to the triggering signal, as only 360 μs is needed per event. However, the limiting factor in the sorting process is the speed at which the droplets can move through the detection zone without compromising their integrity. Therefore, our automatic droplet sorting device achieved a maximum sorting rate of 60 droplet/s.Sorting half of the droplet population produced(i.e.,~500,000 droplets or~50,000 droplets with single cells)takes roughly 2.5 h.
Finally, the third chip enables us to combine those positive-fluorescence droplets with other reagents for further processing or amplification(Figure 5C). The droplet merging device shows a success rate of 95% at a high throughput of 200 droplets/s. Therefore, merging the positive-fluorescence droplets (e.g., sorted out from the previous device)with lysis buffer or amplification reagents can take less than 0.5 h.
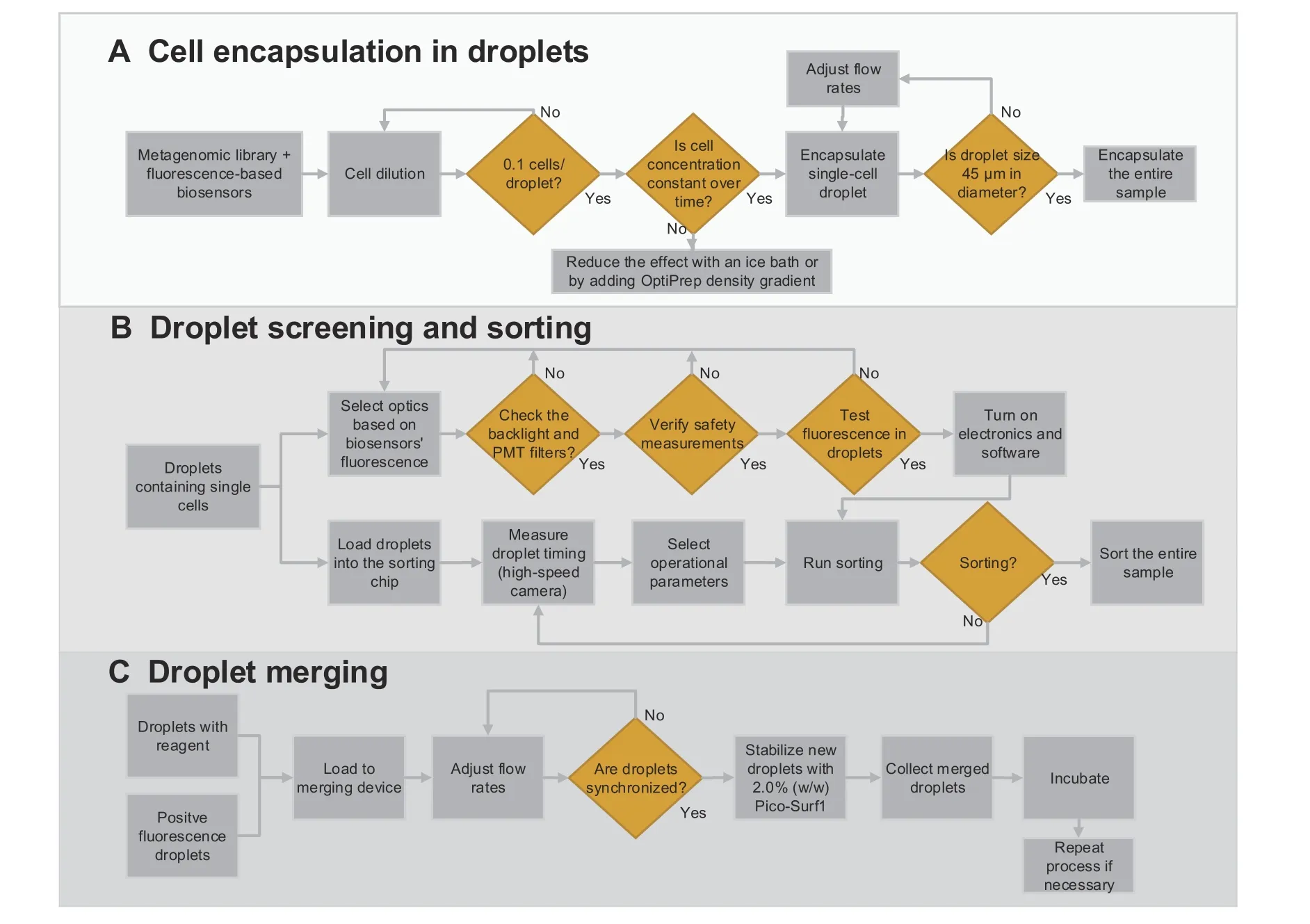
Figure 5 Droplet microfluidic capabilities enabling functional metagenomic studiesThis figure summarizes the three droplet microfluidic tools integrated in this study.A.Process diagram fully describing single-cell encapsulation.A critical step of this procedure is to measure the cell concentration before the encapsulation to guarantee that only one cell is encapsulated per droplet.B.Protocol for droplet sorting.This protocol includes selecting the right optical components and identifying possible errors on the hardware and software.Measuring the time from droplet detection to the sorting junction is fundamental for synchronizing the control system with the device’s droplet flow. C. Merging process of two droplets to add new reagents to already-made droplets.The most crucial step in this process is to adjust the inlet flow rates to obtain a 1:1 droplet synchronization.
Once the metagenomic material is obtained from the positive droplets, the next steps consist of performing sequencing and bioinformatics.
This protocol allows the finding of functional genes out of vast metagenomic libraries. Our group is currently working on a case study that focuses on the metagenomic application of the methods presented in this publication.In this follow-up work,we apply the techniques presented here to search for lipolytic enzymes in Red Sea environmental samples. This metagenomic investigation lead to identifying three novel lipase genes. These genes showed thermostability and high activity based on alignment tests and functional assay results.
Conclusion
A recipe for bulk micromachining high-quality and longlasting master molds on silicon is provided for the first time.Although our procedure requires specialized DRIE equipment, it shows a more robust solution to mold manufacturing than traditional master mold techniques such as SU-8 lithography because these molds can be used indefinitely(e.g.,>100 times)without showing signs of wear.
Three microfluidic droplet techniques for single-cell studies are also described:single-cell encapsulation,fluorescence-based automatic droplet sorting, and droplet merging. For encapsulation, cell concentration is the most crucial parameter,and therefore it must be measured before starting any experiment.A concentration of 0.1 cells/droplet provides the best results to obtain single-cell encapsulation.Our droplet encapsulation system generates droplets at rates of~250–350 droplets/s and effectively encapsulates approximately 25 droplets/s.Automatic droplet sorting is then described entirely, providing guides to optics, instrumentation, and controls. The electronics response time from fluorescence detection to the activation of triggering signal takes only 360 μs per event. However, controlled delays are programmed as we can only move the droplets at~60 droplets/s through the channel without tearing them apart. Our setup is robust and can work with four wavelengths of fluorescent excitation (i.e., 405, 488, 561, and 637 nm), making it compatible with several fluorescenceemitting biosensors. A summary of fluorochromes compatible with this setup is also provided as a quick selection guide.We present a passive droplet merging device that can achieve efficiencies of 95% and at~200 droplets/s. This merger facilitates the addition of precise amounts of reagents for various purposes,such as starting or terminating a reaction and adding new reagents.
Finally,we discuss the potential of these three microfluidic technologies in mining the immense pool of metagenomic resources for bioprospecting.
CRediT author statement
David Conchouso:Methodology, Investigation, Writing -original draft, Writing - review & editing.Amani Al-Ma’abadi:Resources.Hayedeh Behzad:Resources.Mohammed Alarawi:Resources.Masahito Hosokawa:Methodology.Yohei Nishikawa:Methodology.HarukoTakeyama:Methodology.Katsuhiko Mineta:Methodology, Supervision, Writing - review & editing.Takashi Gojobori:Conceptualization, Methodology, Supervision,Writing - review & editing. All authors have read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
The work was supported by the grants from King Abdullah University of Science and Technology (KAUST), Saudi Arabia(Grant Nos.BAS/1/1059/01/01,URF/1/1976/03/01,URF/1/1976-17-01, URF/1/1976-20-01, and FCS/1/3326-01-01). Finally, we thank the nanofabrication and the microfluidics Core Laboratories at KAUST for their support in realizing these microfluidic technologies.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2021.03.010.
ORCID
0000-0002-9788-0977 (David Conchouso)
0000-0001-8895-5423 (Amani Al-Ma’abadi)
0000-0001-7216-8337 (Hayedeh Behzad)
0000-0001-8986-8209 (Mohammed Alarawi)
0000-0002-7070-4690 (Masahito Hosokawa)
0000-0001-5443-7230 (Yohei Nishikawa)
0000-0002-2058-8185 (Haruko Takeyama)
0000-0002-4727-045X (Katsuhiko Mineta)
0000-0001-7850-1743 (Takashi Gojobori)
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Genomics, Proteomics & Bioinformatics
- Call for Papers Special Issue on “Artificial Intelligence in Omics”
- Call for Papers Special Issue on “RNA Modifications and Epitranscriptomics”
- Advanced Single-cell Omics Technologies and Informatics Tools for Genomics, Proteomics, and Bioinformatics Analysis
- A Single-cell Transcriptome Atlas of Cashmere Goat Hair Follicle Morphogenesis
- Polar Gini Curve: A Technique to Discover Gene Expression Spatial Patterns from Single-cell RNA-seq Data