A Single-cell Transcriptome Atlas of Cashmere Goat Hair Follicle Morphogenesis
2021-02-24WeiGeWeidongZhangYuelangZhangYujieZhengFangLiShanheWangJinwangLiuShaojingTanZihuiYanLuWangWeiShenLeiQuXinWang
Wei Ge, Weidong Zhang, Yuelang Zhang, Yujie Zheng, Fang Li, Shanhe Wang,2,Jinwang Liu,Shaojing Tan,Zihui Yan,Lu Wang,Wei Shen,Lei Qu,Xin Wang,
1Key Laboratory of Animal Genetics, Breeding and Reproduction of Shaanxi Province, College of Animal Science and Technology,Northwest A&F University, Yangling 712100, China
2College of Animal Science & Technology, Yangzhou University, Yangzhou 225000, China
3Life Science Research Center, Yulin University, Yulin 719000, China
4College of Life Sciences, Qingdao Agricultural University, Qingdao 266109, China
Abstract Cashmere,also known as soft gold,is produced from the secondary hair follicles(SHFs)of cashmere goats.The number of SHFs determines the yield and quality of cashmere;therefore,it is of interest to investigate the transcriptional profiles present during cashmere goat hair follicle development. However, mechanisms underlying this development process remain largely unexplored,and studies regarding hair follicle development mostly use a murine research model.In this study, to provide a comprehensive understanding of cellular heterogeneity and cell fate decisions, single-cell RNA sequencing was performed on 19,705 single cells of the dorsal skin from cashmere goat fetuses at induction(embryonic day 60;E60),organogenesis(E90),and cytodifferentiation(E120)stages.For the first time,unsupervised clustering analysis identified 16 cell clusters,and their corresponding cell types were also characterized.Based on lineage inference,a detailed molecular landscape was revealed along the dermal and epidermal cell lineage developmental pathways. Notably, our current data also confirmed the heterogeneity of dermal papillae from different hair follicle types, which was further validated by immunofluorescence analysis. The current study identifies different biomarkers during cashmere goat hair follicle development and has implications for cashmere goat breeding in the future.
KEYWORDS Single-cell transcriptome;Cashmere goat;Cellular heterogeneity;Developmental trajectory;Hair follicle morphogenesis
Introduction
Cashmere goat hair follicles can be divided into primary hair follicles (PHFs) and secondary hair follicles (SHFs).Cashmere hair is only produced by SHFs of cashmere goats and the development of SHFs starts during the fetal stage[1,2].The number of SHFs determines the yield and quality of cashmere. Every year, more than 20,000 tons of cashmere is generated in China and,as a consequence,cashmere goats have become an important source of income for farmers who live in North China [3]. It is therefore of interest to decipher the molecular pathways during early hair follicle development in cashmere goats.Studies employing a murine research model have demonstrated that molecular pathways during early hair follicle morphogenesis play important roles in regulating hair characteristics, including fiber length,fineness,and curvature[4].The cashmere goat is different from the mouse,so it is important to reveal the molecular pathways driving cashmere hair follicle morphogenesis. Due to the relatively long duration of pregnancy (around 145–159 days) in cashmere goats [5], our current understanding of their hair follicle morphogenesis is limited.
Similar to the murine model, fetal hair follicle development of the cashmere goats can be divided into three main stages: induction stage (embryonic day 55–65; E55–65),organogenesis stage(E85–95),and cytodifferentiation stage(around E115) [6]. In mice, the molecular foundations underlying the induction stage have recently been comprehensively investigated following the development of singlecell RNA sequencing (scRNA-seq) technology, while the latter two stages remain to be investigated[7].According to studies using the murine model, the formation of placodes and dermal condensates (DCs) are two milestone events during the induction stage that require conserved crosstalk between dermal and epidermal cell populations, including Wnt/β-catenin signaling, Edar signaling, and Fgf signaling[8–10].Mok et al.describe how murine DC formation can be further divided into three substages using scRNA-seq technology, and they characterize detailed gene expression signatures during each substage;they also demonstrate that the extracellular matrix/adhesion signaling is vital for early DC fate commitment [11]. The organogenesis stage is characterized by the formation of dermal papillae (DPs),hair shafts (HSs), and inner root sheaths (IRSs), and the molecular pathways involved includePDGFαsignaling andShhsignaling[12,13].For the cytodifferentiation stage,the differentiation of IRSs, HSs, and keratinocytes becomes obvious and Eda signaling is known to play a role [4,14].An asynchronous development of different hair follicles takes place during the cytodifferentiation stage in mice;this includes guard hair follicles (starting from E13.5), awl/auchene hair follicles (starting from E15.5), and zigzag hair follicles (starting from E17.5) [15]. More recently, we reported a single-cell transcriptome atlas of murine hair follicle morphogenesis using skin tissues from E13.5(induction stage), E16.5 (organogenesis stage), and P0(cytodifferentiation stage),and recapitulated key molecular events during epithelium/dermal cell lineage fate decisions[16].Noteworthy,our murine research also revealed a heterogeneous population of DPs, which provides insight into the asynchronous development of hair follicles. However,the detailed molecular machinery underlying the asynchronous development of different hair follicles remains to be investigated [17,18].
In a preliminarily attempt to reveal the molecular pathways involved in cashmere goat hair follicle morphogene-sis,several research groups have collected skin samples from goat fetuses and performed transcriptome sequencing analysis to reveal gene expression dynamics between different time points [19,20]. However, due to a lack of conserved markers for labelling the particular cell types within hair follicles, most studies have used skin tissues to perform transcriptome sequencing analysis and generated “equali-zed” expression matrices, which sometimes do not reveal the real scenario[21–23].The paucity of information regarding cell heterogeneity within hair follicles has obviously become the main obstacle in deciphering hair follicle morphogenesis. scRNA-seq has recently become a robust tool for dissecting cell heterogeneity, and several groups have also successfully used this technology to reveal the molecular machinery underlying murine hair follicle development [11,16,24], further emphasizing its prospective application in hair follicle development-related research.
To tackle the paucity of information regarding the cellular heterogeneity and molecular pathways underlying key cell fate decisions during cashmere hair follicle development,the current study reported a single-cell transcriptome landscape during cashmere goat hair follicle morphogenesis based on 19,705 single-cell transcriptional profiles. The results provided valuable information for the identification of biomarkers through dissecting cellular heterogeneity during cashmere goat hair follicle development. Furthermore, cell lineage inference analysis provided a comprehensive understanding of the molecular pathways underlying major cell lineage fate decisions, which has implications for future cashmere goat breeding.
Results
scRNA-seq identified different cell types in developing cashmere goat skin
To provide in-depth insight into the molecular profiles during cashmere goat hair follicle development and the main cell fate transitions,skin samples were collected from E60(induction stage),E90(organogenesis stage),and E120(cytodifferentiation stage)of cashmere goat fetuses(Figure1A, Figure S1A), and scRNA-seq was performed on the samples. In total, 7000 single cells were captured and at least 16,000 genes were detected for each sample (Figure S1B). The genome mapping rate was higher than 90% for all samples(Figure S1B).For quality control,the cells were filtered according to the number of genes detected (Figure S1C), and high-quality cells were retained for downstream analysis. After quality control, 19,705 single-cell transcriptome expression profiles were analyzed from the dorsal skin of E60(6825 single cells),E90(6873 single cells),and E120(6007 single cells)cashmere goat fetuses.

Figure 1 scRNA-seq delineated cellular heterogeneity during cashmere goat hair follicle developmentA.Overall experimental design.B.tSNE plot of all single cells labelled with developmental time.Cells from different developmental time points are colorcoded.C.tSNE plot of all single cells labelled with cell types according to their marker gene expression.Different colors represent different cell clusters and the cell number for each cluster is listed in the bracket. D. Dot plot of representative marker genes for different cell clusters. The color intensity represents log1p-transformed expression level,and the dot size represents the positive cell percentage(count>0).E60,embryonic day 60;E90,embryonic day 90; E120, embryonic day 120; DP, dermal papilla; tSNE, t-distributed stochastic neighbor embedding.
To investigate cellular heterogeneity, t-distributed stochastic neighbor embedding (tSNE) analysis was performed,and 16 different cell clusters were identified across three developmental time points (Figure 1B and C; Table S1). By analyzing the cluster-specific expressed genes,different cell types were identified according to their marker gene expression (Figure S1D). Briefly, clusters 1, 4, 6, 7,and 12 expressed high levels of the dermal cell lineage markersLUMandCOL1A1[24], and they were termed as dermalVIM+, dermalDLK+, dermalDKK1+, proliferative fibroblast, and DP, respectively, according to their marker gene expression [17]. For clusters 0, 3, 8, 9, and 11, they expressed high levels of the epithelial lineage markersKRT14,KRT17andKRT85[25,26], and they were termed as epidermalCXCR4+, keratinocyteKRT85+, epithelialKRT14+, keratinocyteKRT71+,and keratinocyteKRT1+,respectively.Furthermore, several important clusters were also identified including HS clusters (LHX2andMSX1, clusters 2 and 5)[27], endothelial cluster (KDRandPECAM1, cluster 10)[28], pericyte cluster (TPM2andACTA2, cluster 13) [29],muscle cell cluster (CNMDandARSI, cluster 15), and macrophage cell cluster(AIF1andRGS1,cluster 14)[30].It was also worth noting that some clusters showed time-dependent accumulation, thus deciphering the process of cell differentiation at a particular time point (Figure S1E). To further verify the cellular heterogeneity revealed by tSNE analysis, we performed consensus clustering analysis of tSNE identified clusters [31]. The results showed that the cells derived from the same lineage were clustered into the same module (Figure S1F), thus further verifying our cellular heterogeneity analysis. More importantly, the transcriptional characteristics for each cell type were delineated,and a series of cell type-specific marker genes were identified during cashmere goat hair follicle development(Figure 1D).It is worth noting that many cell type-specific expressed marker genes were consistent with a murine scenario, such as dermal cell markersPOSTN,DCN, andAPOE, epithelial cell markersKRT14andKRT15, and DP cell markersSOX2andSOX18. Together, we successfully identified different cell types at single-cell resolution and characterized their cell type-specific gene expression patterns,which provide potential biological markers for future research.
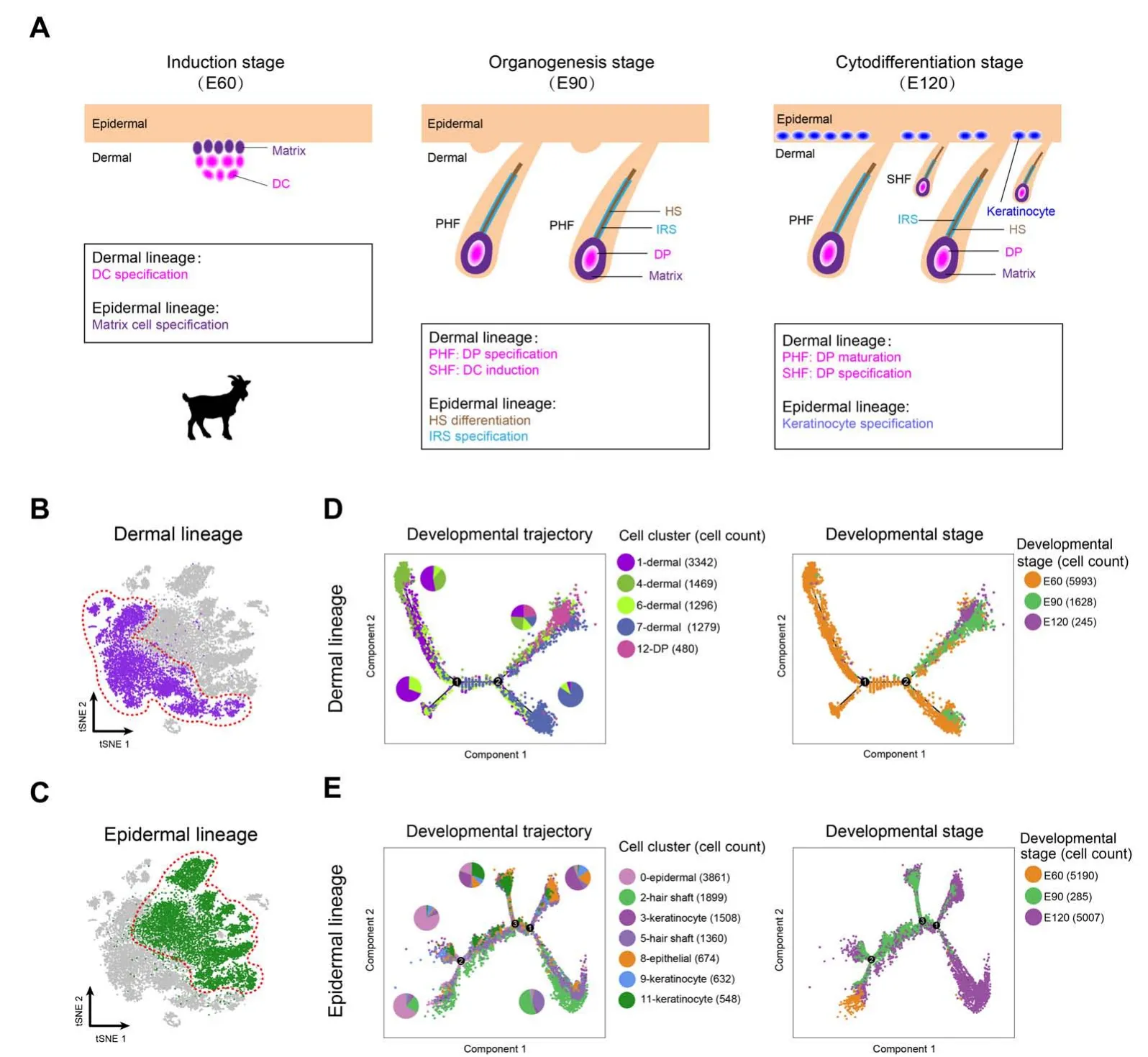
Figure 2 Dermal and epidermal cell lineage developmental trajectories delineated by pseudotime trajectory inference analysesA. A simplified diagram showing the relationship of dermal and epidermal cell lineages at different time points. B. Dermal cell lineage highlighted (red dashed circle) in the tSNE plot. C. Epidermal cell lineage highlighted (red dashed circle) in the tSNE plot. D. Developmental trajectory of dermal cell lineage along pseudotime.Cells were color-coded with cell types identified by Seurat(left panel)and developmental time points(right panel),respectively.The pie chart shows the percentage of each cell cluster for each branch.E.Developmental trajectory of epidermal cell lineage along pseudotime.Cells are color-coded with cell types(left panel)and developmental time points(right panel),respectively.The pie chart shows the percentage of each cell cluster for each branch. DC, dermal condensate; PHF, primary hair follicle; SHF, secondary hair follicle; HS, hair shaft; IRS, inner root sheath.
Recapitulation of cell fate commitment of dermal and epidermal cell lineages based on trajectory inference
After characterizing different cell clusters, the major cell fate transitions were then investigated during hair follicle development. Based on the well-defined anatomical structures of different cell populations and our recent work on constructing murine fetal hair follicle developmental trajectory [7,16], the goat dermal and epidermal cell lineages were then illustrated combining with our tSNE identified clusters (Figure 2A). Pseudotime trajectory construction analysis was then performed on dermal and epidermal cell clusters, respectively (Figure 2B and C). Since all the cell clusters had been successfully characterized, the dermal lineage cell clusters(Figure 2B,clusters 1,4,6,7,and 12)and epidermal lineage cell clusters(Figure 2C,clusters 0,2,3,5,8,9,and 11)were then selected to infer the cell lineage developmental trajectory. For dermal cell lineage, pseudotime trajectory displayed two branch points (Figure 2D),while epidermal cell lineage showed three branch points(Figure 2E). It is worth noting that when the cells were color-coded with their corresponding developmental time points, they also showed a time-ordered pattern along pseudotime. Besides, to further verify the trajectory inference analyses in the dermal and epidermal cell lineages,we performed RNAvelocity analyses because RNAvelocity can be used to infer the developmental directionality by distinguishing the unspliced and spliced mRNAs[32].The results showed that the majority of the RNAvelocity vectors displayed obvious directions toward the branch endpoint(Figure S2A and B), and thus verified our trajectory inference analyses. Altogether, the successful recapitulation of cell fate of dermal and epidermal cell lineages in the current study enabled an in-depth understanding of the key molecular pathways driving cell fate decisions during cashmere goat hair follicle development.
Cross-species comparison between mouse and cashmere goat revealed conserved regulators during DC fate commitment
After dermal cell lineage trajectory inference, we initially focused on the first branch point on the dermal cell pseudotime trajectory to reveal the first dermal cell fate decision.By analyzing gene expression dynamics along pseudotime,2679 differentially expressed genes(DEGs)were observed at the end of cell fate commitment (the gene set 4; Table S2); moreover, gene functional enrichment analysis revealed that these genes were enriched in GO terms of“tissue morphogenesis”, “response to growth factor”, and“cell morphogenesis involved in differentiation” (Figure3A).A comparison of overlapping GO terms between each gene set showed that a substantial number of GO terms were shared (Figure S3A and B). Of particular note, a series of cashmere goat orthologs of canonical murine DC markers were observed in these DEGs,includingAPOD,LUM,andAPOE[11]. Furthermore, the expression levels of DC markers, includingAPOD,SOX18,CTNNB1, andSOX2,were increased along pseudotime (Figure 3B). Therefore,we termed the first branch point as DC fate commitment.
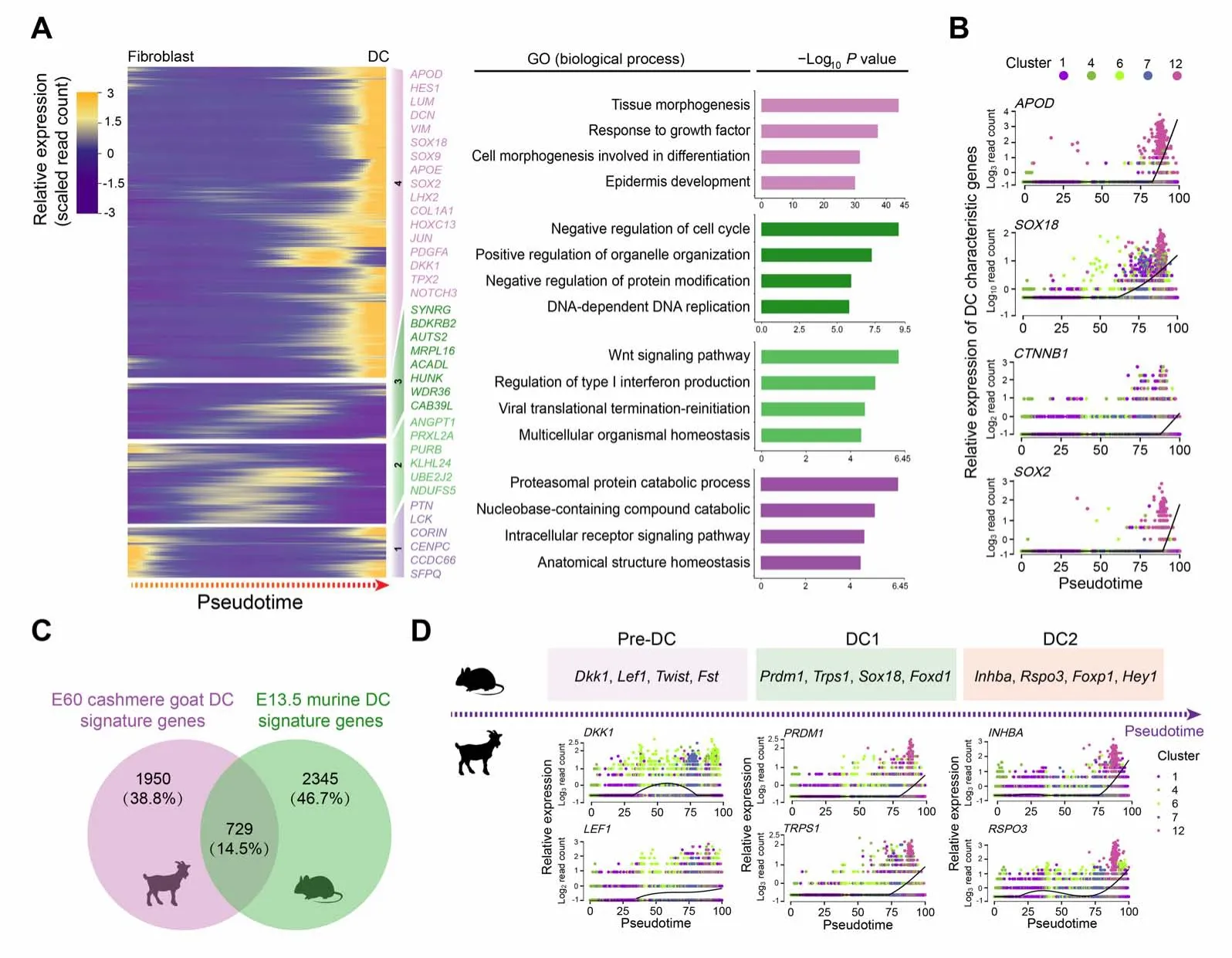
Figure 3 Pseudotime trajectory analysis delineated molecular profiles during DC fate commitmentA.Pseudotime expression heatmap during DC fate commitment.The four gene sets were determined by k-means clustering according to their expression patterns, and the top 4 GO terms for each gene set are listed in the panel on the right. B. Visualization of cashmere goat orthologs of murine DC characteristic genes along pseudotime. Cells are color-coded according to the cell clusters as shown on the top panel. C. Venn diagram illustrating overlapping characteristic genes between E60 cashmere goat DC cells and murine E13.5 DC cells.D.Visualization of cashmere goat orthologs of murine DC characteristic genes at different stages along pseudotime.Murine characteristic DC markers are shown in the top panel and the expression patterns of their orthologs in the cashmere goat are shown in the lower panel. Cells are color-coded according to the cell clusters as shown on the right. E13.5,embryonic day 13.5; GO, Gene Ontology; Pre-DC, precursor of dermal condensate; DC1, dermal condensate stage 1; DC2, dermal condensate stage 2.
To our knowledge, no reports to date have delineated cellular heterogeneity and transcriptional landscape during cashmere goat hair follicle morphogenesis,and most studies regarding hair follicle development have employed a murine model. To gain an in-depth insight into the machinery driving DC fate commitment, we then compared DC fate characteristic genes with recently reported murine DC fate commitment characteristic genes [11] (Figure 3C); an overlap of 729(14.5%)genes was observed(Table S3).It is interesting to note that, based on scRNA-seq on E15.0 dorsal skin, Mok et al. recently demonstrated that murine DC fate commitment could be divided into pre-DC, DC1,and DC2 stages [11]. After analyzing the cashmere goat orthologs of murine DC marker genes at different stages,we also found that characteristic DC genes in cashmere goats also showed chronological expression patterns along pseudotime (Figure 3D). Briefly, the expression of cashmere goat orthologs of murine pre-DC markers,DKK1andLEF1,was elevated prior to the expression of DC1 and DC2 markers along pseudotime, such asPRDM1,TRPS1,INHBA, andRSPO3. It is therefore plausible that DC fate commitment in cashmere goats may also involve several different stages. Together, these findings indicate that DC fate commitment might require a highly conserved regulatory network during hair follicle development.
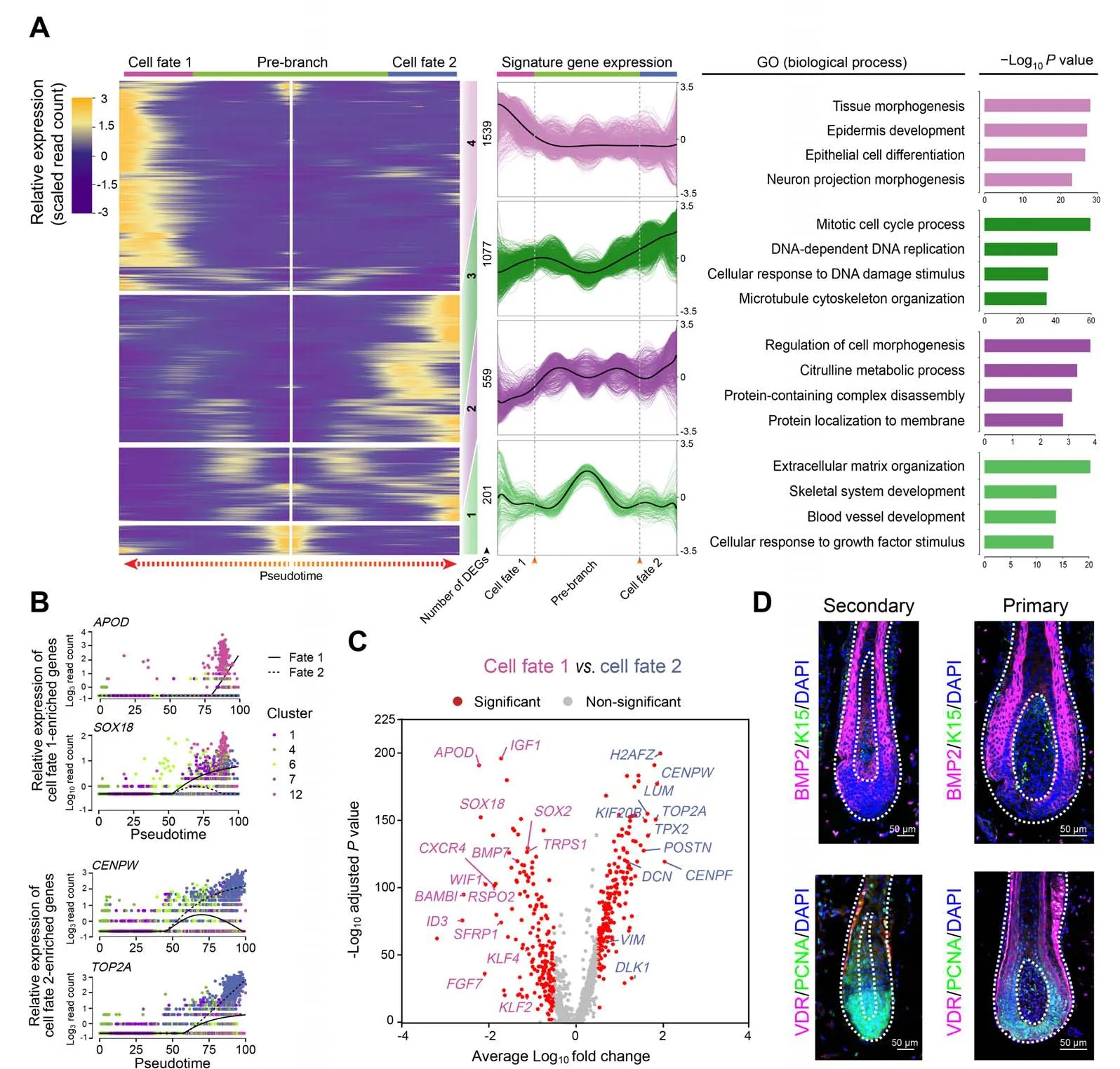
Figure 4 DPs from PHFs and SHFs in the cashmere goat showed distinct gene expression profilesA.Heatmap illustrating dynamic gene expression profiles during DP cell fate commitment.The gene expression pattern for each gene set is shown in the middle panel, and the top 4 enriched GO terms for each gene set are listed in the right panel. B. Pseudotime expression patterns of cell fate 1-enriched genes APOD and SOX18 and cell fate 2-enriched genes CENPW and TOP2A.Cells are color-coded according to the cell clusters as shown on the right.C.Volcano plot illustrating cell fates 1 and 2 as well as cell fate significantly enriched DEGs. D. Immunofluorescence analysis of BMP2, K15, VDR, and PCNA in PHFs and SHFs from E120 cashmere goat skin sections. Scale bar = 50 μm. DEG, differentially expressed gene; BMP2, bone morphogenetic protein 2; K15, keratin 15; VDR, vitamin D receptor; PCNA, proliferating cell nuclear antigen; DAPI, 4′,6-diamidino-2-phenylindole.
Primary and secondary hair follicles showed distinct gene expression signatures
After defining DC fate commitment,we focused on the next branch point, which mainly involved the heterogeneity of DPs. Noteworthy, knockout ofSox18mainly affected the development of zigzag hair follicles in mice,thus providing evidence that the unsynchronized development of hair follicles requires different signaling pathways[33].Consistent with such a hypothesis, our recent research on mice also revealed that guard/awl/auchene DPs and zigzag DPs were transcriptionally distinct [16]. To verify whether the unsynchronized development of goat PHFs and SHFs also involves distinct molecular pathways in goat DPs,the DEGs between the two branches were compared.This comparison revealed that cell fate 1 elevated canonical DP marker genes(such asAPODandSOX18) enriched in the GO terms of“tissue morphogenesis” and “epidermis development”,while cell fate 2 elevated genes (such asCENPWandTOP2A) enriched in the GO terms of “mitotic cell cycle process” and “DNA-dependent DNA replication” (Figure4A and B, Figure S4A and B). To gain an in-depth understanding of the differences between the two branches, we identified DEGs using another differential analysis method,the Wilcoxon rank-sum test. Consistent with Monocle analysis, cell fate 1 also showed higher expression ofAPOD,SOX18,andIGF1,while cell fate 2 showed elevated expression ofCENPW,TOP2A,andDCN(Figure 4C).GO enrichment analysis similarly revealed that DEGs in cell fate 1 were enriched in the GO terms of “tissue morphogenesis”and“epithelial cell differentiation”,while DEGs in cell fate 2 were enriched in the GO terms mainly related to the regulation of cell cycle process (Figure S4C).
To dissect the heterogeneity within DPs, we performed immunofluorescence analysis on the dorsal skin of E120 cashmere goat fetuses and observed different staining patterns between PHFs and SHFs (Figure 4D). A previous study has demonstrated that bone morphogenetic protein(BMP)signaling regulates the size of hair follicles in mice[34]; we, therefore, compared BMP2 expression between PHFs and SHFs in goat.The results showed that BMP2 was specifically expressed in the matrix cells surrounding DPs in the PHFs,while BMP2-positive cells were found in both DPs and surrounding matrix cells in the SHFs. Moreover,consistent with our previous finding that the SHFs showed higher expression of cell cycle-related genes (TOP2A,CENPF,CENPW,etc.)[35],our analysis also revealed that proliferating cell nuclear antigen (PCNA), a cell cycle-related marker, was expressed mainly in matrix cells surrounding DPs in PHFs,while was highly expressed in both DPs and surrounding matrix cells in SHFs.Taken together,it is plausible that different DP signals induce the development of different hair follicle types in the cashmere goat.
Delineating the transcriptional characteristics and developmental pathways during the first epidermal cell fate decision
After revealing dermal cell fate decision,we focused on the epidermal cell clusters. Pseudotime trajectory inference analysis by Monocle revealed that epidermal cells showed three different branch points. Based onk-means clustering and DEG dynamics analysis along pseudotime,four distinct gene clusters were classified at the first branch point. As expected, a series of matrix cell markers were observed at the end of pseudotime, includingHOXC13,KRT25, andKRT71, thus deciphering a matrix cell fate commitment process (Figure 5A). GO enrichment analysis showed that matrix cell-expressed characteristic genes were enriched in the GO terms of “keratinocyte differentiation”, “epidermis development”, and “skin epidermis development” (Figure 5B, Figure S5A), while a comparison of the GO terms between different gene sets revealed that gene sets 1 and 2 differed from those of gene sets 3 and 4(Figure S5B).
To gain an in-depth insight into molecular profiles during matrix cell fate commitment,characteristic gene expression patterns along pseudotime were further analyzed.For gene set 4,namely matrix cell markers,a series of keratin family genes were enriched,includingKRT25,KRT27,andKRT84,and their expression increased along pseudotime (Figure 5C). For gene sets 3 and 2, we found transiently elevated expression ofLEF1,SBSN,SOX18, andSOX9, and enrichment of the GO terms of “cardiac epithelial to mesenchymal transition”and“mesenchymal cell differentiation”for gene set 3 and“skin development”and “morphogenesis of an epithelium”for gene set 2(Figure 5B and C).For gene set 1, genes such asVIM,LUM,POSTN, andCOL1A1were enriched and all showed decreased expression along pseudotime (Figure 5C). Immunofluorescence analysis also confirmed that LEF1 and CTNNB1 were expressed in the upper epidermis,which was consistent with a murine scenario (Figure 5D) [36,37]. Collectively, immunofluorescence analysis here verified our single-cell transcriptome analysis results, and cell fate trajectory inference analysis here offered valuable insights for understanding the molecular pathways underlying the underappreciated epidermal cell development.
Increased transcriptional divergence between mouse and cashmere goat during HS and IRS cell fate commitment
After deciphering matrix cell fate commitment at the first epidermal trajectory point, we focused on the next branch point.By analyzing DEGs along pseudotime,we found that cell fate 1 enriched canonical HS markers,includingSHH,VDR, andHOXC13, while cell fate 2 enriched canonical IRS markers such asSOX9,KRT14,SBSN, andELF1(Figure 6A) [27]; immunohistochemistry results further confirmed the expression of HOXC13 and SOX9 in cashmere hair follicles (Figure S6A). For pre-branch,LUM,COL1A1,OGN, andSOX18showed decreased expression along pseudotime(Figure 6B).For HS cell fate(cell fate 1),elevated expression ofDCN,TOP2A,PCNA, andH2AFZdisplayed elevated expression and enriched in the GO terms“mitotic cell cycle process”and“DNA replication”(Figure S6B and C).For IRS cell fate(cell fate 2),PRDM1,KRT1,SOX9, andKRT14were increased expression along pseudotime (Figure 6B) and enriched in the GO terms of “supramolecular fiber organization”and“tissues morphogenesis”(Figure S6B and C).
In addition, the identified HS and IRS characteristic genes were compared with the recently identified murine HS and IRS characteristic genes[16].Interestingly,we only observed an~8.8% overlap in IRS characteristic genes between mice and cashmere goats (Figure 6C; Table S4),and these mainly consisted of keratin family genes,such asKRT14,KRT17,andKRT79[38].Further comparison of the top 20 expressed transcriptional factor genes between mice and cashmere goats revealed four conserved transcriptional factor genes, includingKLF3,KLF4,KLF5, andBARX2(Table S4), of which, theKlffamily members (Klf3/4/5)were three identified key regulons during mouse IRS fate commitment as we previously reported in our mouse singlecell data [16]. At the same time, the overlap in HS characteristic genes between mice and cashmere goats was about 4.5% (Figure 6D; Table S4), includingMSX1,HOXC13,APOO,BMP4,LHX2,SHH, andVDR[39–41]. To further validate our analyses, the expression of PCNA and VDR were compared between E90 cashmere goat skin and E16.5 murine skin tissues; immunohistochemistry revealed similar expression patterns during hair follicle development(Figure 6E). These data together demonstrated that HS and IRS specifications may employ a conserved program during cell fate decisions. Noteworthy, the decrease of the overlapping characteristic genes along pseudotime was also consistent with recent findings that the similarity of organspecific expressed developmentally dynamic genes decreased along the developmental process[42].
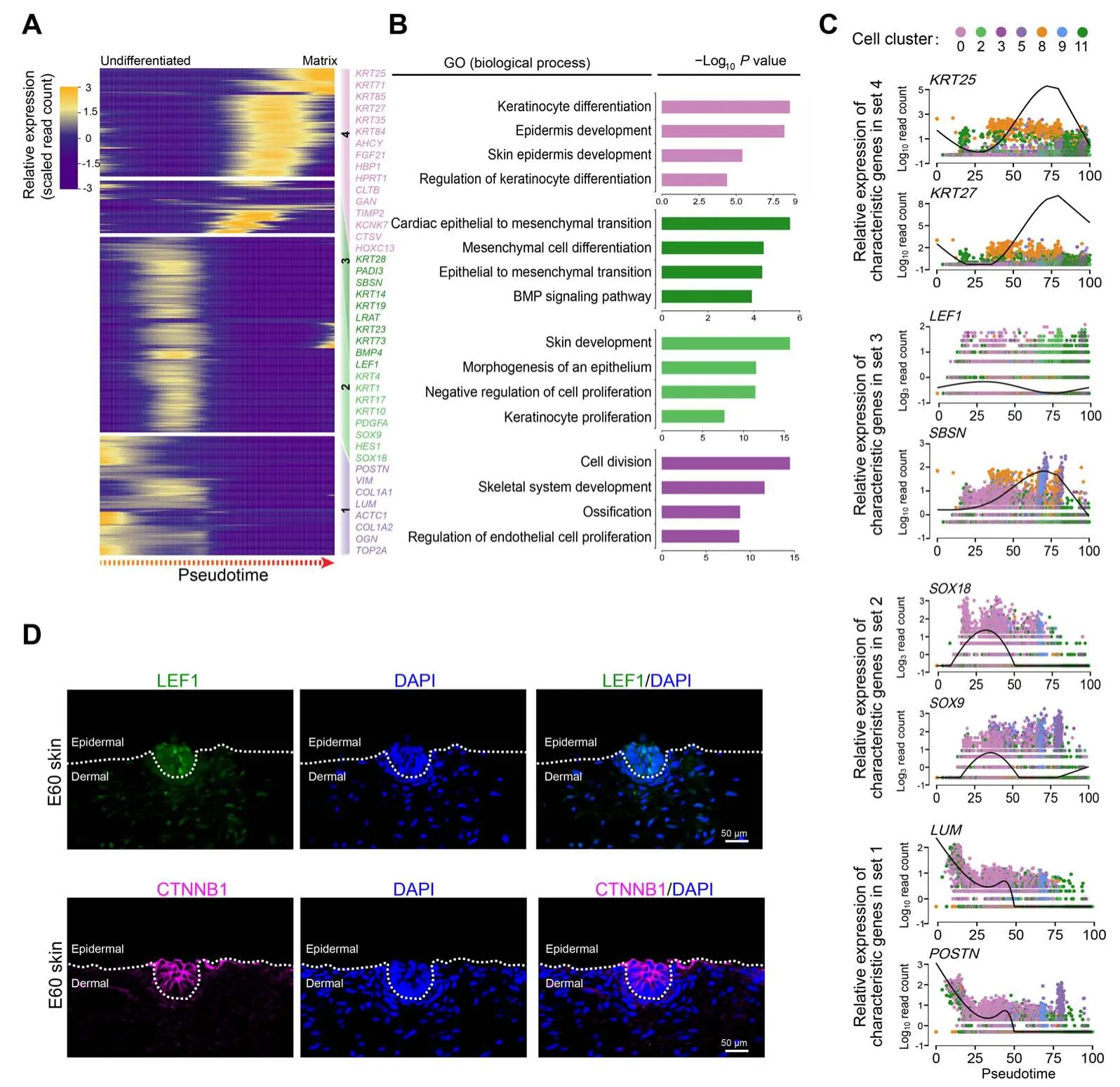
Figure 5 Delineating matrix cell fate decision along pseudotimeA.Heatmap demonstrating dynamic gene expression patterns during matrix cell fate commitment.DEGs were divided into four different gene sets based on k-means clustering. B. GO enrichment analysis of characteristic genes in the four different gene sets shown in (A). C. The pseudotime expression pattern of representative characteristic genes from each gene set. Cells are color-coded according to the cell clusters as shown atop. D. Immunofluorescence analysis of LEF1 and CTNNB1 in E60 cashmere goat skin tissues.Scale bar=50 μm.LEF1,lymphoid enhancer-binding factor 1;CTNNB1,catenin beta 1.
Revealing the developmental pathways during keratinocyte cell fate commitment
After delineating the first two epidermal cell lineage cell fate decisions, the last branch point was explored.Pseudotime trajectory analysis also revealed two different branches. Pseudotime gene expression dynamics were subsequently analyzed between the two branches (Figure7A). When analyzing the DEGs along pseudotime, we found that cell fate 1 enriched genes such asVDR,BMP4,STAR,KRT85,andKRT14,while cell fate 2 enriched genes such asBMP2,SHH,CUX1,andETV5.It is of interest thatKRT14has been identified as a marker for keratinocytes both in humans and mice[26,43].We,therefore,termed cell fate 1 as“keratinocytes”.To gain in-depth insight into their corresponding cell types, we performed gene functional enrichment analysis and pseudotime GO enrichment analysis for each gene set(Figure 7B,Figure S7A).The results showed that for pre-branch,DEGs were mainly enriched in terms of“regulation of response process”,while for cell fate 1, DEGs were mainly enriched in terms of “epidermal cell differentiation”. Interestingly, for cell fate 2, DEGs were mainly enriched for “regulation of cell cycle” with lower expression ofKRT14(Figure 7C, Figure S7B). It is therefore plausible that keratinocyte differentiation at this stage was not synchronized. To confirm this hypothesis, immunofluorescence analysis of VDR, PCNA, BMP2,CTNNB1,and LEF1 was performed,and the results showed that the expression of VDR and BMP2 in the outer layer of the epidermis was not homogeneous,with some cell clusters showing higher expression while others showing lower expression(Figure7D).The pre-branch enriched CTNNB1 was uniformly expressed in the interfollicular epidermis.Furthermore, the expression patterns of PCNA and LEF1 were more obvious and they were partially expressed in the epidermis. Taken together, these results emphasized that keratinocyte differentiation in cashmere goats was asynchronous and required different gene expression profiles.
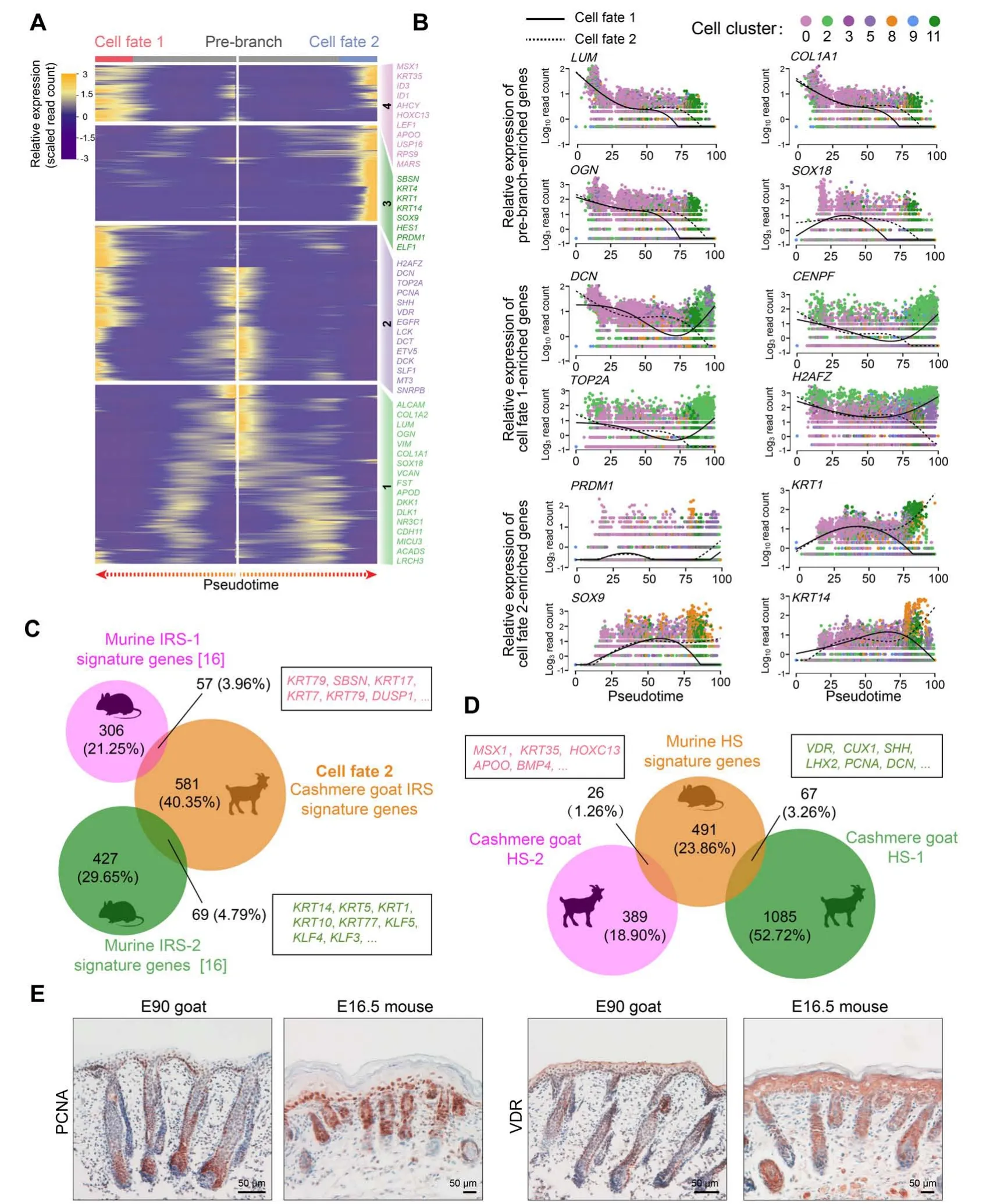
Figure 6 Cross-species comparison of the HS and IRS signature genes revealed increased divergence during hair follicle developmentA.Heatmap illustrating the pseudotime gene expression pattern of DEGs during IRS and HS cell development.Cell fate 1 depicts HS fate,while cell fate 2 depicts IRS fate.B.Pseudotime expression patterns of representative marker genes during IRS and HS cell fate commitment.Cells are color-coded according to the cell clusters as shown atop; the solid line depicts cell fate 1, while the dashed line depicts cell fate 2. C. and D. Venn diagram demonstrating overlapping IRS(C)and HS(D)characteristic genes between mouse and cashmere goat.The representative overlapping genes are listed in the corresponding boxes.E.Comparison of PCNA and VDR expression in E90 cashmere goat and E16.5 mouse skin tissues.Scale bar=50 μm.E16.5,embryonic day 16.5.
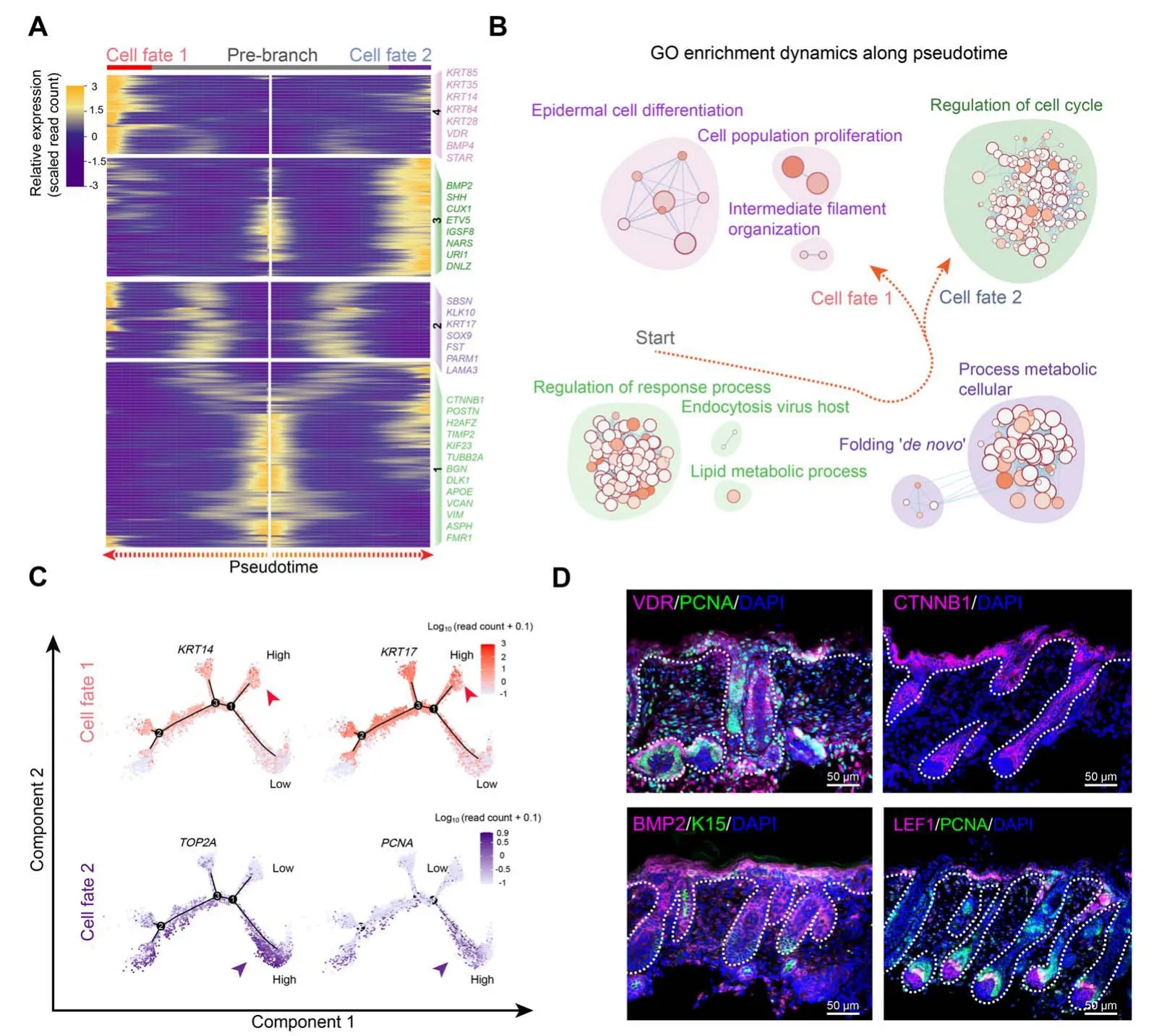
Figure 7 Dissection of the asynchronous development of keratinocytesA.Heatmap illustrating gene expression dynamics during keratinocyte differentiation.B.GO terms corresponding to the gene sets shown in(A).Arrows indicate the elongation of pseudotime.C.Expression of cell fate 1 characteristic genes KRT14 and KRT17,and cell fate 2 characteristic genes TOP2A and PCNA along the pseudotime trajectory. D. Immunofluorescence analysis of VDR, PCNA, CTNNB1, BMP2, LEF1, and K15 in E90 cashmere goat skin tissues. Scale bar = 50 μm.
Discussion
scRNA-seq is a robust tool for investigating organogenesis and, in recent years, has provided us with unparalleled insights into mammalian development[44,45].Over the past decade, the number of papers using scRNA-seq based technology has increased exponentially and the Science magazine also announces scRNA-seq technology as the Breakthrough of the Year 2018 [46,47]. Here, we successfully constructed a single-cell atlas to reflect cashmere goat hair follicle development. Based on the downstream analysis, we revealed unparalleled insights into the cellular heterogeneity and major cell fate decisions taking place during in utero cashmere goat hair follicle morphogenesis.As far as we know,this is the first study to comprehensively delineate the molecular profiles of various cell types and reveal major cell fate decisions during hair follicle development in this species.Our study here also provides evidence that different DP signals may participate to orchestrate the development of different hair follicles in cashmere goats.More importantly, by analyzing cluster-specific gene expression profiles, the current data provide a valuable resource for future studies in cashmere goat skin tissues and an in-depth understanding of follicle development.
In the current study, 19,705 single-cell transcriptional profiles were analyzed from three different developmental stages, which could represent major cell types during hair follicle development. To dissect cellular heterogeneity during cashmere goat hair follicle development after tSNE analysis, cluster-specific expressed characteristic genes were analyzed for each cluster to infer their corresponding cell types.It is worth noting that all the cell markers used in the current study were referenced from the murine model due to the paucity of information regarding marker gene expression during hair follicle development in cashmere goats. However, the current study showed that substantial murine cell type-specific biomarkers were identical to those in the cashmere goat. Furthermore, by comparing the cell type-specific characteristic genes between the cashmere goats and mice(Figures 3C,6C,and 6D),we found that the transcriptome similarity decreased along pseudotime (as revealed by the overlapping of gene signatures in this study). Briefly, about 729 DC characteristic genes were found to be overlapped between cashmere goats and mice,and murine DC characteristic genes at different stages(pre-DC,DC1,and DC2)showed similar expression patterns to those along pseudotime in the cashmere goat [11]. Meanwhile,for the IRS and HS cell populations in the late stage of hair follicle development,the percentage of overlapping genes decreased (126 and 93 overlapping genes, respectively). Similarly, by comparing gene expression from seven different species across the brain, heart,ovary, kidney,testis,and liver tissues,researchers have demonstrated that organ-specific molecular profiles are more similar during early development and become more distinct during later stages of development [42]. It is, therefore, plausible that hair follicle development in cashmere goats and mice may also be consistent with the aforementioned theory.
Similar to findings in the murine model [16], we also observed differential gene expression profiles in DP populations from different hair follicles (PHFsvs. SHFs).Moreover, an in-depth comparison of DEGs revealed that different DP populations required different transcriptional profiles. Therefore, it is plausible that different induction signals may be involved during the asynchronous development of hair follicles[48].Consistent with such a hypothesis,SOX2was found to be specifically expressed in guard hair follicles but not in zigzag hair follicles, whileSOX18was demonstrated to regulate zigzag hair follicle morphogenesis [17,33,49]. Although our data here delineated the asynchronous development of different hair follicles in cashmere goats, the detailed machinery underlying such a phenomenon was not investigated.Future studies might focus on such topics,which could provide us with new insights into hair follicle biology and hair follicle regeneration.
Based on single-cell pseudotime trajectory inference,the current study also highlighted previously underappreciated cell fate decisions during cashmere goat hair follicle development. Different from murine epidermal cell lineage trajectory showing only two branch points,the pseudotime lineage trajectory of epidermal cell lineage in cashmere goats showed three different branch points. By analyzing the gene expression profiles of the first two branch points,similar cell fate decisions were shown,while the additional branch point in cashmere goats mainly came from keratinocyte differentiation. Such difference in pseudotime trajectory may be partially explained by differences in the development of hair follicles across species. For example,E120 stage cashmere goats show obvious hair fibers on the surface of the skin,while is not until post-natal day 6–7 that the hair follicles in murine skin become visible. The difference in the pseudotime trajectory revealed that hair follicle development is heterochronous between cashmere goats and mice; this is commonly found when comparing the development of specific organs across species.Besides,it is also noteworthy that RNA velocity analysis revealed two groups of cells with opposite velocity vectors.Whether this is an indicator of convergence in cellular differentiation remains to be investigated in future studies.
Finally, the current dataset provided an important resource for understanding the cellular heterogeneity and major cell fate decisions during cashmere goat hair follicle development.To our knowledge,it is the first time that the detailed transcriptional landscape of different cell populations at a single-cell resolution has been described, which makes it a valuable resource for the identification of biomarkers in the future. In addition, by dissecting DP cell heterogeneity,our study emphasized that distinct DP signals orchestrate the asynchronous development of different hair follicle types.Furthermore,our trajectory inference analysis successfully recapitulated major cell fate decisions during cashmere goat hair follicle development,which enabled us to comprehensively study detailed developmental pathways involved in hair follicle morphogenesis. These findings together provide us with new insights into cashmere goat hair follicle biology and will have implications for cashmere goat breeding and animal husbandry.
Materials and methods
Experimental animals
All the experimental Shanbei White cashmere goats involved in this study were obtained from the Shanbei cashmere Goat Engineering Technology Research Center of Shaanxi Province (Yulin, China) and were fed with cashmere goat feeding standard (DB61/T583-2013) of Shaanxi Province. All pregnancies were initiated by artificial insemination with standard procedures.
Single-cell suspension preparation
Goat fetuses at the desired dates were delivered using caesarean section after the pregnant goats had been anaesthetized with xylazine hydrochloride (Catalog No.070011582,Huamu Animal Health Products Co.Ltd.,Jilin,China).Tissue blocks(0.5 cm×0.5 cm)were resected from the dorsal skin of each fetus and were immediately transferred to ice-cold DMEM/F12 media (Catalog No.C11330500BT, Gibco, Beijing, China) with 50 U/ml penicillin and 50 mg/ml streptomycin(Catalog No.SV30010,HyClone, Logan, UT). After washing three times with DMEM/F12 to remove any blood cells, the skin tissues were then dissociated into single cells ready for sequencing. For the E60 and E90 samples, the obtained skin tissues were firstly incubated with 2 mg/ml collagenase IV(Catalog No. C5138, Sigma, St Louis, MO) for 30 min at 37°C, and then mechanically dissociated into single-cell suspensions using a 1-ml pipette tip.For the E120 samples,the skin tissues were firstly cut into~3 mm pieces and then incubated with 2 mg/ml collagenase IV for 30 min at 37°C. After incubation, the hair follicles within the skin tissues were isolated using a pair of precise forceps and the pooled hair follicles were further dissociated into single cells with TypLE Express (Catalog No. 12605028, Gibco,Grand Island, NY) for 30 min at 37°C. The obtained single-cell suspensions were then washed three times with phosphate-buffered saline(PBS)supplemented with 0.04%bovine serum albumin (BSA; Catalog No. V900933, Sigma)and were filtered with a BD Falcon 40-μm cell strainer(Catalog No. 352340, BD Biosciences, San Jose, CA) to remove debris and cell aggregations. For each stage, the samples were obtained from at least two different goat fetuses, and for each fetus, a single-cell suspension was prepared separately until finally pooled together before single-cell barcoding.
Single-cell library construction and sequencing
A single-cell library was constructed using a 10X Genomics’Chromium Single Cell 3′V3 Gel Beads Kit(Catalog No.PN-1000075,10X Genomics,Pleasanton,CA)according to the manufacturer’s instructions. After cell counting,the single-cell suspension was adjusted to 1000 cells/μl,and about 7000 cells were obtained for each stage. The singlecell barcoding procedure was performed using a 10X Genomics Chromium barcoding system (10X Genomics,Pleasanton, CA) according to the manufacturer’s guide.After single-cell library construction, an Illumina HiSeq X Ten sequencer (Illumina, San Diego, CA) was employed,and 150-bp pair ended reads were generated.
10X Genomics scRNA-seq data processing
The obtained raw sequencing files were processed with the standardCellRanger(v2.2.0)pipeline according to the 10X Genomics official guide (https://www.10xgenomics.com/cn/). The produced raw base call files were firstly transformed intofastqfiles using theCellranger mkfastqfunction. The goatARS1reference genome downloaded from Ensembl was used as a reference genome (https://asia.ensembl.org/Capra_hircus/Info/Index). TheCellranger countfunction was used to perform mapping, filtering of lowquality cells, barcode counting, and unique molecular identifier (UMI)counting.
After the standardCellRangerpipeline, the generated gene expression matrice files were analyzed using theSeurat(V2.3.4) package according to the official user guidelines(https://satijalab.org/seurat/vignettes.html).Quality control was performed using theFilterCellsfunction, and cells with detected genes < 200 and genes with detected cells<3 were deprecated.After normalization and data scaling,the different datasets were integrated using theRunMultiCCAfunction.tSNE was used to perform dimension reduction analysis and different cell clusters were identified using theFindClustersfunction. Those clusters consisting of specifically expressed genes were analyzed with theFindAllMarkersfunction and with the parameter“min.pct=0.25,thresh.use=0.25”.
Unsupervised clustering analysis
Unsupervised clustering analysis was performed using single-cell consensus clustering(SC3)[31].All procedures used in the current study were performed following the online tutorial (https://www.bioconductor.org/packages/release/bioc/vignettes/SC3/inst/doc/SC3.html#sc3-input).SC3 requires theSingleCellExperimentobject as input, we therefore firstly transformed theSeuratobject into theSingleCellExperimentobject using the “as.Single-CellExperiment”function.Other parameters were set using the default parameter.
Single-cell pseudotime lineage trajectory reconstruction
Single-cell lineage reconstruction analysis was performed using theMonocle(v2) package according to the online tutorial (http://cole-trapnell-lab.github.io/monocle-release/docs/).TheMonocleobject was constructed from theSeuratobject using thenewCellDataSetfunction, andSeuratdetermined variable genes were used as ordering genes to order cells in pseudotime along a trajectory. Dimension reduction was performed usingDDRTreemethods. To analyze differential gene expression between different cell branches, theBEAMfunction was used, and DEGs were identified withqvalue < 10−4. A branch-specific gene expression heatmap was plotted with theplot_genes_branched_heatmapfunction, and different gene sets were calculated according tok-means clustering.
RNA velocity analysis
RNA velocity analysis was performed according to the officialvelocyto.Rtutorial (https://github.com/velocytoteam/velocyto.R). Briefly, the unspliced and spliced reads were firstly generated fromCellRangergenerated bam files usingvelocyto.pypipeline. After that,velocyto.Rwas utilized to calculate the velocity vectors,and the RNA velocity vectors were embedded into themonocleobject using the“show.velocity.on.embedding.cor”function.
Immunofluorescence analysis and immunohistochemistry staining
Immunofluorescence analysis and horseradish peroxidase(HRP)-based immunohistochemistry staining were performed as previously described [50,51]. For immunofluorescence analysis, the paraffin-embedded skin tissues were firstly deparaffinized in xylene and further rehydrated in ethanol solution. Antigen retrieval was performed in 0.01 M sodium citrate buffer at 96°C. After a permeabilization procedure in 0.5 M Tris-HCI buffer supplemented with 0.5% Triton X-100 (Catalog No.T8200, Sorlabio, Beijing, China) for 10 min, the slides were then blocked with 3% BSA and 10% donkey serum(Catalog No. AR0009, Boster, Wuhan, China) in 0.5 M Tris-HCI buffer for 30 min. Primary antibodies diluted in the blocking buffer were incubated with the slides at 4°C overnight, and then corresponding secondary antibodies were added and incubated at 37°C for 1 h.4′,6-diamidino-2-phenylindole (DAPI) was used to stain the nuclei. The pictures were taken using a Nikon AR1 confocal system(Nikon, Tokyo, Japan). For HRP-based immunohistochemistry staining, following antigen retrieval,the samples were firstly incubated with 3% H2O2for 10 min at room temperature to remove endogenous peroxidase activity. Primary antibodies were added and incubated with samples at 4°C overnight;the corresponding HRP-labeled secondary antibodies were then added and incubated for 40 min at room temperature. Subsequently,the peroxidase substrate 3,3-diaminobenzidine (DAB;Catalog No.ZLI-9017,Zsbio,Beijing,China)was used to produce a chromogenic reaction with hematoxylin to stain the nuclei. The slides were finally mounted with neutral resins,and pictures were captured with an Olympus BX51 microscope imaging system(Olympus,Tokyo,Japan).All the primary and secondary antibodies used in this study are listed in Table S5.
Ethical statement
All the experimental procedures involving goats were reviewed and approved by the Experimental Animal Management Committee of Northwest A&F University, Yangling, China.
Data availability
The cashmere goat scRNA-seq data used in this study have been deposited in the Genome Sequence Archive[52]at the National Genomics Data Center, Beijing Institute of Genomics,Chinese Academy of Sciences/China National Center for Bioinformation (GSA: CRA002671) which are publicly accessible at https://ngdc.cncb.ac.cn/gsa, and also in the Gene Expression Omnibus repository at the National Center for Biotechnology Information (GEO: GSE144351) which are publicly accessible at https://www.ncbi.nlm.nih.gov/geo/.
CRediT author statement
Wei Ge:Methodology, Formal analysis, Visualization,Software, Conceptualization, Writing - original draft,Writing - review & editing.Weidong Zhang:Methodology, Formal analysis, Visualization.Yuelang Zhang:Methodology,Formal analysis,Visualization.Yujie Zheng:Formal analysis,Validation,Visualization.Fang Li:Formal analysis, Visualization.Shanhe Wang:Formal analysis,Visualization.Jinwang Liu:Formal analysis, Validation,Resources.Shaojing Tan:Formal analysis, Visualization,Software.Zihui Yan:Formal analysis, Validation, Visualization, Software.Lu Wang:Formal analysis, Validation,Visualization, Software.Wei Shen:Supervision, Writing -review & editing.Lei Qu:Supervision, Resources.Xin Wang:Conceptualization,Supervision,Writing-review&editing. All authors have read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This study was supported by the National Natural Science Foundation of China(Grant Nos.31972556 and 31772573).We are grateful to Prof. Lei Qu and Dr. Jinwang Liu (Life Science Research Center,Yulin University,China)for their kind help regarding cashmere goat breeding and surgical procedures during sample preparation.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2021.07.003.
ORCID
0000-0002-4610-7933 (Wei Ge)
0000-0003-0363-0086 (Weidong Zhang)
0000-0002-7922-3834 (Yuelang Zhang)
0000-0002-0345-9066 (Yujie Zheng)
0000-0002-3342-9394 (Fang Li)
0000-0003-2888-7515 (Shanhe Wang)
0000-0001-7174-0004 (Jinwang Liu)
0000-0002-7465-9946 (Shaojing Tan)
0000-0002-5491-6810 (Zihui Yan)
0000-0002-7517-8140 (Lu Wang)
0000-0001-6112-2501(Wei Shen)
0000-0001-7167-879X (Lei Qu)
0000-0002-2845-8510 (Xin Wang)
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- scDPN for High-throughput Single-cell CNV Detection to Uncover Clonal Evolution During HCC Recurrence
- Mapping Human Pluripotent Stem Cell-derived Erythroid Differentiation by Single-cell Transcriptome Analysis
- Single-cell Long Non-coding RNA Landscape of T Cells in Human Cancer Immunity
- Single-cell Transcriptomes Reveal Characteristics of MicroRNAs in Gene Expression Noise Reduction
- Single-cell RNA Sequencing Reveals Sexually Dimorphic Transcriptome and Type 2 Diabetes Genes in Mouse Islet β Cells
- Single-cell RNA Sequencing Reveals Thoracolumbar Vertebra Heterogeneity and Rib-genesis in Pigs