Mapping Human Pluripotent Stem Cell-derived Erythroid Differentiation by Single-cell Transcriptome Analysis
2021-02-24ZijuanXinWeiZhangShangjinGongJunweiZhuYanmingLiZhaojunZhangXiangdongFang
Zijuan Xin,Wei Zhang,Shangjin Gong,Junwei Zhu,Yanming Li,Zhaojun Zhang,3,4,5,, Xiangdong Fang,3,4,5,
1CAS Key Laboratory of Genome Science and Information,Beijing Institute of Genomics,Chinese Academy of Sciences/China National Center of Bioinformation, Beijing 100101, China
2College of Life Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
3Sino-Danish College, University of Chinese Academy of Sciences, Beijing 100190, China
4Institute for Stem Cell and Regeneration, Chinese Academy of Sciences, Beijing 100101, China
5Beijing Key Laboratory of Genome and Precision Medicine Technologies, Beijing 100101, China
Abstract There is an imbalance between the supply and demand of functional red blood cells (RBCs) in clinical applications.This imbalance can be addressed by regenerating RBCs using several in vitro methods.Induced pluripotent stem cells(iPSCs)can handle the low supply of cord blood and the ethical issues in embryonic stem cell research,and provide a promising strategy to eliminate immune rejection.However,no complete single-cell level differentiation pathway exists for the iPSC-derived erythroid differentiation system.In this study,we used iPSC line BC1 to establish a RBC regeneration system. The 10X Genomics single-cell transcriptome platform was used to map the cell lineage and differentiation trajectory on day 14 of the regeneration system. We observed that iPSC differentiation was not synchronized during embryoid body(EB)culture.The cells(on day 14)mainly consisted of mesodermal and various blood cells,similar to the yolk sac hematopoiesis. We identified six cell classifications and characterized the regulatory transcription factor (TF)networks and cell–cell contacts underlying the system. iPSCs undergo two transformations during the differentiation trajectory, accompanied by the dynamic expression of cell adhesion molecules and estrogen-responsive genes. We identified erythroid cells at different stages, such as burst-forming unit erythroid (BFU-E) and orthochromatic erythroblast(ortho-E) cells, and found that the regulation of TFs (e.g., TFDP1 and FOXO3) is erythroid-stage specific. Immune erythroid cells were identified in our system.This study provides systematic theoretical guidance for optimizing the iPSCderived erythroid differentiation system, and this system is a useful model for simulating in vivo hematopoietic development and differentiation.
KEYWORDS scRNA-seq;iPSC;Hematopoiesis;Erythropoiesis;Differentiation trajectory
Introduction
The substantial gap between the supply and demand for blood has always been a serious clinical practice problem[1].Artificial blood is an essential means of solving this worldwide shortage, and obtaining functional red blood cells (RBCs) is the key to artificial blood generation [2].RBC regenerationin vitrohas been an important research direction in the blood research field for many years [3,4].Currently,the primitive materials that can be used for RBC regeneration are mainly cord blood hematopoietic stem cells (HSCs) [5], embryonic stem cells (ESCs) [6,7], and induced pluripotent stem cells (iPSCs) [6,8]. The regeneration technology of cord blood HSCs is relatively mature; however,some problems come with this technology, such as difficulty in obtaining materials, significant differences among individuals, and the high price of this technology. ESCs are subject to ethical restrictions [9].Human iPSCs can be differentiated into various cell typesin vitro,providing a model for basic research and a source of clinically relevant cells[10–12].Although the derivation of iPSC lines for patients has now become a routine technique,how to accurately differentiate iPSCs into the desired cell types is still a major challenge[13,14].
Decades of research have shown thatin vitrohematopoietic differentiation is closely related toin vivodevelopment[15].Hematopoiesis originates from the FLK1/KDR+lateral mesoderm, and some of the specialized cells can be induced to form hemangioblasts by the transcription factor(TF) ETV2 [16,17]. These cells can differentiate into the blood and vascular precursor cells, during which ETV2 is regulated by the BMP and WNT signaling pathways[18,19]. At this time, the primitive RBCs that express embryonic globin (ε-globin; encoded byHBE) are produced,which is the first wave of hematopoiesis in the yolk sac[20].Hemangioblasts differentiate into endothelial cells and are further specialized into hematopoietic-endothelial cells(HECs) that produce erythroid-myeloid progenitors(EMPs). EMPs can differentiate into most of the myeloid cells and into definitive RBCs that can simultaneously express embryonic globin (ε-globin; encoded byHBE), fetal globin(γ-globin;encoded byHBG1andHBG2), and adult globin (β-globin; encoded byHBB); this differentiation represents the second wave of hematopoiesis in the yolk sac[21]. HSCs can also be directly produced through the endothelial-hematopoietic transition (EHT) induced by GATA2 and RUNX1 [22–24]. This occurs in the aorta-gonad-mesonephros(AGM)region that maintains the lifelong hematopoietic differentiation and the hematopoietic cycle[25], and these cells can differentiate into definitive RBCs that express only adult globin and are regulated by signaling pathways such as VEGF and HIF [26,27]. Hematopoietic differentiation is the differentiation process of HSCs to form various types of blood cells [28]. After the differentiation and activation of HSCs,it is necessary to go through the cell fate determination program regulated by GATA1, KLF1,and other TFs to enter the erythroid differentiation process[29]. The generation of RBCs is an elaborate multistep process involving the differentiation of early erythroid progenitor cells into definitive RBCs, which requires spatiotemporal-specific completion of globin synthesis and assembly[30,31],iron metabolism[32,33],heme synthesis[34], cell denucleation, and other processes, eventually forming intact functional RBCs [35]. However, the molecular and cellular mechanisms involved in these processes are still poorly understood.
Based on the existing molecular mechanisms of hematopoietic development and erythroid differentiation,scientists simulate embryonic hematopoiesis [36] using the spin embryoid body (EB) method to regenerate hematopoietic stem progenitor cells (HSPCs)in vitroand then induce the differentiation of RBCs from iPSCs.However, the differentiation efficiency and denucleation rate are low, and adult globin expression is limited; these drawbacks limit the clinical availability [37]. Simultaneously, iPSC differentiation is an elaborate process, and flow cytometry and immunostaining have been used to determine the cell types during iPSC differentiation culture.However,these methods are limited by the number of fluorescent probes used, and it is not possible to solve important problems, such as the cell composition and the differentiation path of the iPSC differentiation system under high-resolution conditions. Recently, single-cell transcriptome technology has been able to capture and identify rare and transient cell types, determine the spatial or temporal localization of cells,and reconstitute gene regulatory networks,helping scientists understand the mechanisms by which development and cell fate are determined [38]. In recent years, there have been many single-cell hematopoiesisstudiesin vitro[39,40].By single-cell sequencing of EBs,researchers found that naïve H9 ESCs have a stronger hematopoietic capacity than primer H9 ESCs [41] and illuminated the heterogeneity of pluripotent stem cell-derived endothelial cell differentiation[42].Single-cell sequencing of day 29 erythrocytes belonging to the iPSC-derived erythroid differentiation system revealed that cells expressing adult globin showed reduced transcripts encoding ribosomal proteins and increased expression of ubiquitin-proteasome system members [43]. The lack of a complete single-cell transcriptome map, as iPSCs differentiate into RBCs, does not allow scientists to guide iPSCs to produce functional RBCs. Therefore, in this study, we established an iPSC-derived erythroid differentiation system and obtained a dynamic transcriptional map of cell differentiation through high-resolution single-cell transcriptome sequencing. The cell map of thein vitroiPSC-derived erythroid differentiation system and thein vitrodifferentiation trajectory of the intact iPSCs to RBCs were mapped for the first time.iPSCs undergo two transformations. During the first transformation (i.e., EHT), iPSCs withdraw from pluripotency into lateral plate mesoderm and differentiate into HECs and then into HSPCs,accompanied by expression fluctuation of cell adhesion molecules. During the second transformation,HSPCs differentiate into erythroid progenitor cells which further differentiate into definitive RBCs. Moreover, estradiol promotes erythroid progenitor cell proliferation and inhibits erythroid differentiation. This study has an important guiding significance for analyzing the basic fate of cells and the gene network structure, as well as for designing more effective hematopoietic and erythropoietic strategies for regenerative medicine.
Results
iPSC-derived HSPC production and erythropoiesis
Depending on the spin EB culture method, we used the iPSC line BC1 to collect the suspension cells(SCs)through the cell strainer in the system after 14 days of EB culture[8,44,45] (Figure 1A). The established culture system can effectively produce SCs. Cell count analysis showed that the number of SCs on day 14 was approximately 9 folds that of iPSCs on day 0 (Figure 1B). Flow cytometry analysis showed that approximately 25.90% ± 3.05% of the SCs expressed cell surface marker CD34,approximately 27.08%± 1.63% of the SCs expressed cell surface marker CD45,and approximately 22.90%of the SCs expressed both CD34 and CD45,indicating that we collected a certain percentage of HSPCs from the SCs (Figure 1C). To detect the hematopoietic differentiation potential of the SCs,we performed colony-forming unit (CFU) assays on the collected SCs from day 14.After two weeks of culture,differential blood lineage colonies were observed and counted under the microscope(Figure 1D and E). It suggested that the SCs have the potential to differentiate into various blood lineage cells.Subsequently,we attempted to induce HSPCs in SCs toward erythroid differentiation using serum-free medium(SFM)[8].By assessing changes in expression of the erythrocyte markers CD71 and CD235a at different time points(Figure 1F),we found that on day 14 of the EB stage,the proportion of CD71 and CD235a double-positive cells in SCs exceeded 30%,indicating that a portion of cells in the system entered the erythroid differentiation stage. On day 28, more than 95%of cells expressed bothCD71 and CD235a(Figure 1F).qPCR assay showed that the expression ofHBBandHBGincreased significantly during the entire system differentiation (Figure 1G). Wright-Giemsa staining analysis showed that most cells were at the terminal erythroid differentiation stage and some cells were denucleated RBCs(Figure 1H). In summary, we successfully constructed an integratedin vitroiPSC-derived erythroid differentiation system by spin EB technology using the iPSC line BC1,as well as obtained and induced HSPCs to enter the erythroid differentiation process, eventually producing denucleated RBCs. Though we have constructed an erythroid differentiation system, we still do not understand what kind of molecular changes of the system’s cells have undergone during the series of processes from pluripotent stem cells to erythroid cells and what factors regulate the process of molecular changes experienced by cells. Therefore, to clarify the aforementioned problem, we collected SCs that contain various cell types on day 14 at the middle time point of the differentiation system and performed single-cell transcriptome sequencing.
Cell composition of the iPSC-derived erythroid differentiation system
To construct a scRNA-seq library,we isolated and selected EB cells (EBCs) and SCs on day 14, and constructed a scRNA-seq library based on a 10X Genomics platform.After rigorous quality control analysis of the data by Seurat and the removal of double cell droplets using R package DoubletFinder[46,47],we obtained 3660 cells from batch 1 and 6538 cells from batch 2,given 10,198 high-quality cells for downstream analysis (Figure S1A; Table S1). Totally,we detected 69,024 informative genes and 1,197,985,924 unique molecular identifiers (UMIs) from two batch samples (Table S1). The correlation analysis of the expression pattern indicated that our two batch samples were positively correlated (Spearman coefficient = 0.904,P< 0.001)(Figure S1B). We then clustered cells based on Seurat’sknearest neighbor method, annotated cells based on R package SingleR [48] and published datasets [49,50], and visualized cells based on uniform manifold approximation and projection(UMAP)[51](Figure 2A and B,Figures S2 and S3).
We classified cells into six categories from the cells belonging to starting cells or coming from different germ layers. iPSCs and ESCs were defined as starting cells (n=1705); astrocytes, keratinocytes, melanocytes, neuroepithelial cells, and neurons were classified as ectodermderived cells [52,53] (n= 1529); epithelial and mesangial cells were classified as intermediate mesoderm-derived cells [54,55] (n= 413); adipocytes, chondrocytes, fibroblasts, hepatocytes, myocytes, mesenchymal stem cells,skeletal muscle cells,smooth muscle cells,and tissue stem cells were classified as paraxial mesoderm-derived cells[56,57] (n= 1934); endothelial cells and pericytes were classified as lateral plate mesoderm-derived cells[58](n=1561); CD34+HSPCs, pro-myelocytes, myelocytes, macrophages, monocytes, neutrophils, eosinophils, CD8+T cells, burst-forming unit erythroid (BFU-E) cells, colonyforming unit erythroid (CFU-E) cells, erythrocytes, and orthochromatic erythroblast (ortho-E) cells were classified as hematopoietic cells(n=3056)(Figure 2C,Figure S4A).

Figure 1 Identification of iPSC-derived HSPCs and erythroid cellsA.Schematic diagram of the protocol used to generate HSPCs and erythroid cells from iPSCs in vitro.B.Histogram showing the counts of iPSCs on day 0 and SCs collected on day 14 per round-bottom 96-well plate.Error bar represents SD.C.Percentage of CD34+and/or CD45+cells in the total SCs(mean±SD).D.Count of colonies growing from SCs collected on day 14.E.The morphology of hematopoietic CFUs growing from SCs.Scale bar=1000 μm.F.Flow cytometry results showing cells expressing CD71 and CD235a on day 14,17,23,and 28 during the erythroid differentiation of SCs.G.qPCR of βchain and γ-chain globin expression on day 4,day 14+3,and day 14+9 during the entire system differentiation.H.The morphology of erythroid cells on day 28.Scale bar=20 μm.iPSC,induced pluripotent stem cell;HSPC,hematopoietic stem progenitor cell;EB,embryoid body;SC,suspension cell;SFM,serum-free medium;BMP4, bone morphogenetic protein 4;FGF-2,fibroblast growth factor-2;SCF,stem cell factor; VEGF, vascular endothelial growth factor; EPO, erythropoietin; CFU, colony-forming unit; IL3, interleukin 3; CFU-E, colony-forming unit erythroid; CFU-G, colony-forming unit granulocyte; CFU-GM, colony-forming unit granulocyte macrophage; CFU-M, colony-forming unit macrophage.
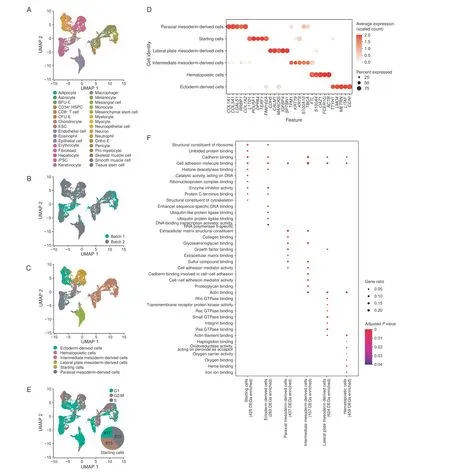
Figure 2 Cell composition in the iPSCs-derived systemA.UMAP visualization of EBCs and SCs.Cells were labeled by SingleR annotation results.B.UMAP plot of cells labeled by batches.C.UMAP plot of cells labeled by cell classifications based on starting cells or cells derived from different germ layers. D. The dot plot of signature genes for each cell classification.E.UMAP visualization of cell cycle analyzed by Seurat.The pie chart showed the percentage of cells in different cell cycles in the starting cells.F.GO term enrichment results of signature genes from each cell classification.EBC,embryoid body cell;BFU-E,burst-forming unit erythroid;ESC,embryonic stem cell; ortho-E, orthochromatic erythroblast; UMAP, uniform manifold approximation and projection; DEG, differentially expressed gene.
To prove the reliability of the cell classification results,we identified the signature genes of each cell classification and calculated the Spearman coefficient between different cell classifications(Figure 2D,Figure S4B).It appears that the expression pattern of each cell classification was specific. We also performed cell cycle analysis based on the expression pattern of genes using Seurat and visualized the cell cycle analysis results into the UMAP dimension reduced map.We noted that among 10,198 cells,2390 cells were in the S phase,2076 cells were in the G2/M phase,and 5732 cells were in the G1 phase (Figure 2E). Specifically,we found that the cell cycle pattern was highly heterogeneous in different cell classifications, especially more than three-quarters of the starting cells in the G2/M and S phases(the pie chart in Figure 2E).Based on the signature genes of six cell classifications, we performed the Gene Ontology (GO) enrichment analysis using R package clusterProfiler to gain insight into the biological processes involved in the EB formation and cell differentiation process[59]. It is impressive that cell adhesion-related pathways were present in all cell classifications (Figure 2F).
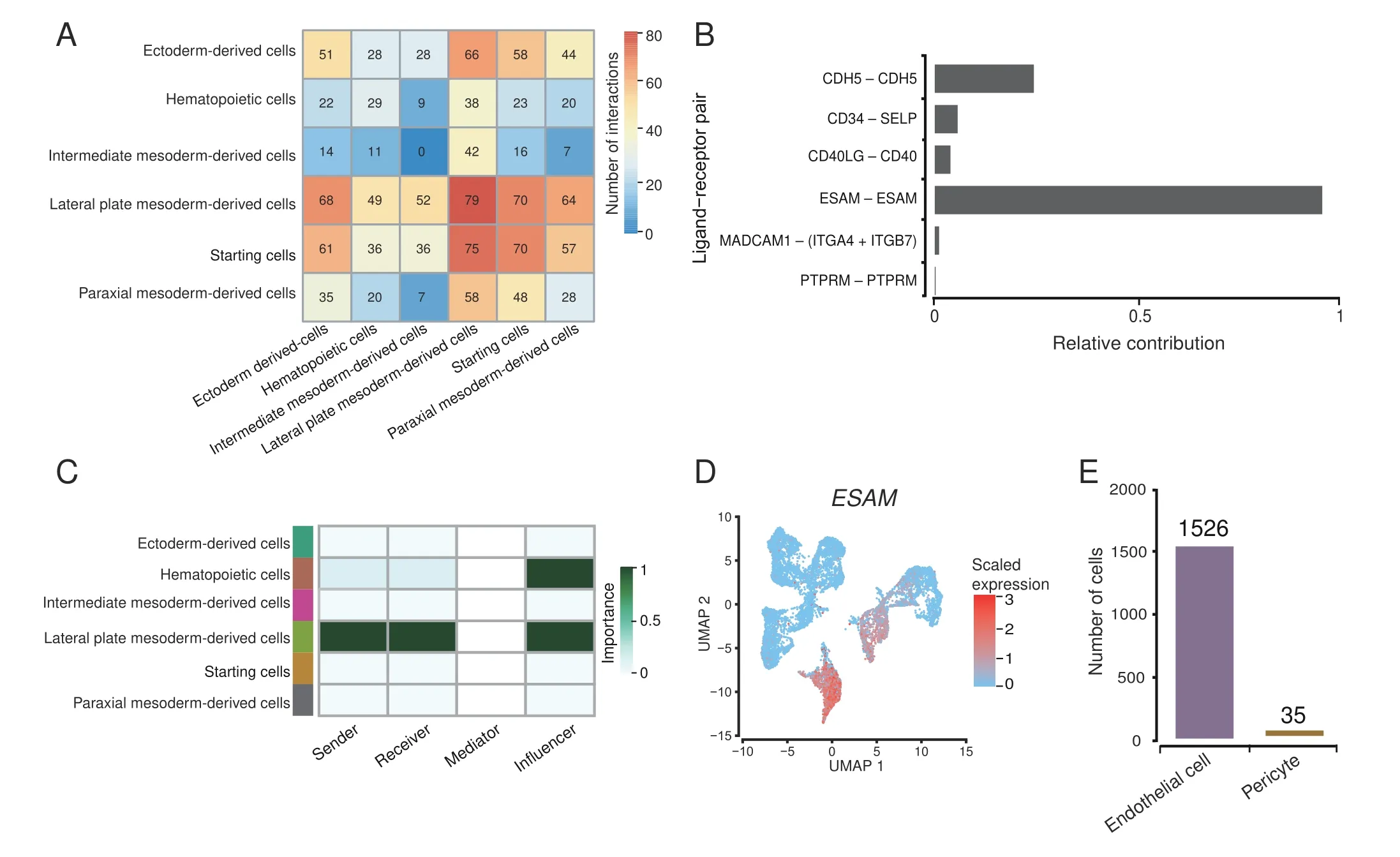
Figure 3 Cell–cell contact analysisA. Heatmap of cell–cell contact scores analyzed by CellChat. B. Contribution of each ligand–receptor pair in CDH5, CD34, CD40, ESAM, MADCAM,and PTPRM signaling pathways.C.The role of each cell classification in ESAM signaling pathway.D.UMAP plot of every single-cell ESAM expression level. E. Bar plot showing cell counts of endothelial cells and pericytes in lateral plate mesoderm-derived cells. CDH5, cadherin 5; SELP, selectin P;CD40LG, CD40 ligand; ESAM, endothelial-specific adhesion molecule; MADCAM1, mucosal addressin cell adhesion molecule 1; ITGA4, integrin subunit alpha 4; ITGA7, integrin subunit alpha 7; PTPRM, protein tyrosine phosphatase receptor type M.
Endothelial-specific adhesion molecule affects hematopoiesis through cell–cell interactions
Considering that cell adhesion-related pathways may play an essential regulatory role during EB formation to hematopoiesis, we next analyzed the mode of cell adhesion between cells in our system.We used CellChat[60]to perform cell–cell contact analysis and evaluate the regulatory function of cell adhesion-related pathways in our system.We found that the contact score between the lateral plate mesoderm-derived cells and other cells were the highest(Figure 3A), and the most significant cell–cell contact signal corresponding to the lateral plate mesoderm-derived cells was the contact between endothelial-specific adhesion molecule (ESAM) ligand and ESAM receptor (Figure 3B,Figure S5A and B). ESAM pathway is an autocrine signaling pathway;it is expressed and secreted by lateral plate mesoderm-derived cells and then acted on itself,and plays a particular regulatory role in hematopoietic cells(Figure 3C and D). The lateral plate mesoderm-derived cells were mainly composed of endothelial cells(Figure 3E).Previous studies have shown that the expression of ESAM enhances the tight junctions between arterial endothelia,which is not conducive for producing HSCs [61]. We performed bulk RNA-seq on SCs collected at day 14 and the iPSC line BC1, and compared the data with published HSPC transcriptome data from human cord blood[62].We found that SCs are significantly different from iPSCs, while SCs display similar hematopoietic lineage-related gene expression patterns to HSPCs and exhibit HSPC-like characteristics (Figure S5C and D). Compared with BC1, SCs also display down-regulated expression of cell adhesionrelated genes(Figure S5E),which may be the direct cause of the separation of SCs from EBs, and the necessary demand for hematopoiesis.
Regulon networks in the iPSC-derived erythroid differentiation system
To clarify the networks of regulons(i.e.,TFs and their target genes) involved in the process from EB formation to hematopoiesis in our system,we used the TFs included in the detected data to reduce dimension and mapped the SingleR cell annotation results to the new TF dimension atlas.The R package,SCENIC,was used to calculate the area under the curve (AUC) scores of regulons [63]. The mapping data showed that the cell differentiation trajectory revealed by TFs is consistent with the clustering by signature genes(Figure 4A),and cells of the same cell classification are still clustered together.According to the AUC scores,regulons’activity has significant cell classification specificity(Figure 4B). Based on regulons’ activity pattern, we constructed the interaction networks of cell classification-specific regulons based on Cytoscape.We observed that part of the target genes of different TFs that were activated in the same cell classification were overlapped, suggesting that the TFs’ regulation was synergistic in our system(Figure 4C).We performed the GO enrichment analysis for target genes in regulons by the clusterProfiler(Figure S6A−P)and mapped the activation positions of regulons in the UMAP atlas(Figure S7).Specifically,we observed that the regulons MECOM, ETS1, ELK3, ERG, and SOX18 showed high activities in lateral mesoderm-derived cells(Figures S6I, S6J, and S7), while the regulons GATA1,TAL1, RUNX1, CEBPD, MYB, and ELF1 were activated in hematopoietic cells(Figures S6K,S6L,and S7).CEBPD and MYB were mainly activated in myeloid cells and targeted to myeloid cell differentiation and cytokine production-related biological processes (Figures S6M, S6N, and S7). GATA1 and TAL1 were activated explicitly in erythroid cells and participated in erythrocyte differentiation,erythrocyte homeostasis, Ras signaling transduction, and GTPase activity regulation(Figures S6O, S6P,and S7).At the same time, RUNX1 and ELF1 exhibited activities in both myeloid and erythroid cells and enriched in cell activation and leukocyte proliferation.
To identify TFs specific to the differentiation stage during hematopoietic differentiation, we calculated the regulon specificity score (RSS) for each cell type included in hematopoietic cells. We noticed that erythroid cells at different stages have a unique RSS pattern,suggesting that the regulation of TFs was erythroid-stage specific (Figure S8). Especially in the erythrocyte cluster, we observed TFDP1 with the highest RSS that can heterodimerize with E2F proteins to control the transcriptional activity of numerous genes involved in cell cycle progression from G1 to S phase [64]. Wang et al. showed that thekyomutant of TFDP1 could cause changes in the morphology and number of erythroid cells during the development of fish embryos,indicating that TFDP1 can be linked to the development of erythroid cells[65].However,the regulatory role of TFDP1 in the development of mammalian erythroid cells has not been clarified. We speculate that TFDP1 may be related to cell number restriction during the maturation of erythroid cells.
Cell differentiation trajectory of the iPSC-derived erythroid differentiation system
To further determine the cell differentiation trajectory during the differentiation of the entire system, we used Monocle2 to order single cells and constructed an entire lineage differentiation trajectory with a tree-like structure[66] (Figure 5A). The results showed that cell differentiation originated from starting cells.After differentiation into different germ layers, two branches appeared. The hematopoiesis branch was mostly composed of hematopoietic cells,and the non-hematopoiesis branch mainly consisted of ectoderm-derived, intermediate mesoderm-derived, lateral plate mesoderm-derived, and paraxial mesoderm-derived cells(Figure 5B,Figure S9A).We use a pseudo-time branch heatmap to show branch-specific expression patterns along two developmental pathways (hematopoiesisvs. non-hematopoiesis). Specifically, we identified 264 signature genes (cluster 1) for pre-branch, including 39 TF coding genes (such asPOU5F1andSOX2), which were mainly enriched in glycolysis/gluconeogenesis; 300 signature genes (cluster 2) were identified for the hematopoiesis branch,includingRUNX1,GATA2,and 21 other TF coding genes which drive pre-branch cells to enter the hematopoiesis branch(cell fate 1),and these signature genes were mainly enriched in the hematopoietic cell lineage pathway;409 signature genes(cluster 3)were identified for the nonhematopoiesis branch, includingSOX7,SOX17, and 41 other TF coding genes which drive pre-branch cells to enter the non-hematopoiesis branch (cell fate 2), and these signature genes were mainly enriched in vascular smooth muscle contraction pathway(Figure 5C and D,Figure S9B).Several research teams have proved that RUNX1 and GATA2 mark the beginning of hematopoiesis [23,67].SOX7 activatesCDH5promoter and hinders the binding of RUNX1 to DNA, and SOX17 mediates the repression of RUNX1 and GATA2, thereby hindering the transition to hematopoietic fate and maintaining the endothelial-arterial identity[67–69].
Meanwhile,Monocle2 divides cells into three states,with pre-branch, hematopoiesis, and non-hematopoiesis branches corresponding to states 1, 2, and 3, respectively(Figure 5E). Analysis of the expression trend of signature genes of each branch in different state cells showed that signature genes of each pseudo-time branch are highly expressed in its corresponding state cells (Figure 5F). The functional annotation analysis showed that state 1 cells(pre-branch) were enriched in the mitotic spindle, G2/M checkpoint, and mitotic prometaphase, indicating that they were in an active cell division state and was regulated by P53, MYC, and mTOR pathways. State 2 cells (hematopoiesis branch) were enriched in heme metabolism,neutrophil degranulation, and other blood cell-related pathways and were regulated by PI3K and IL6.State 3 cells(non-hematopoiesis branch)were enriched in angiogenesis,myogenesis, extracellular matrix organization, and other mesodermal and mesenchymal cell-related pathways, and regulated by signaling pathways, such as TGF-β and hypoxia (Figure 5G, Figure S9C).
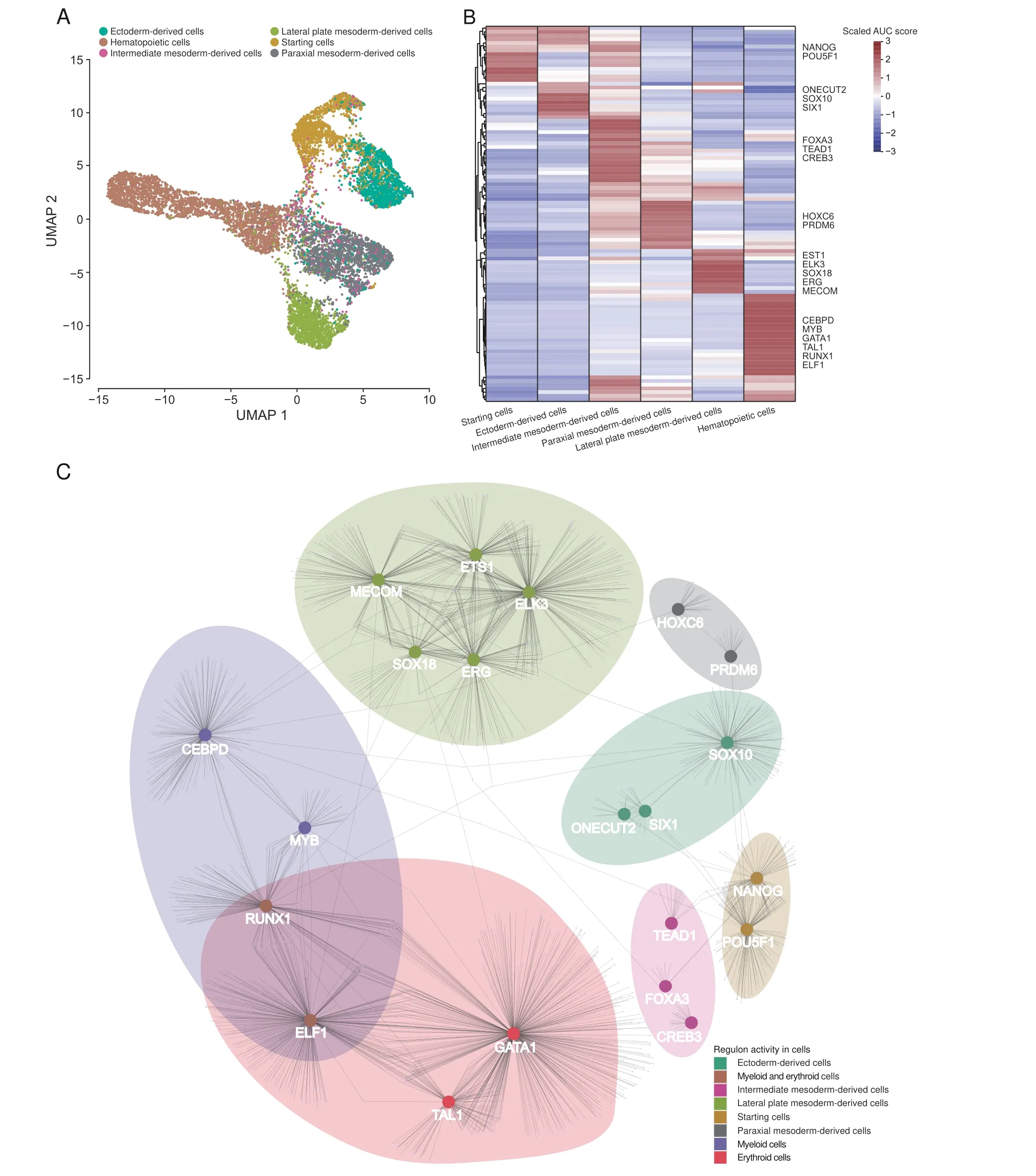
Figure 4 Regulon regulation analysisA. UMAP map of all cells based on human TFs. Colors indicate six cell classifications. B. Heatmap of AUC patterns of regulons related to cell classifications.The regulons with high AUC scores in each cell classification are listed on the right side of the heatmap. C.The network of the regulons with high AUC scores in each cell classification. TF, transcription factor; AUC, area under the curve.
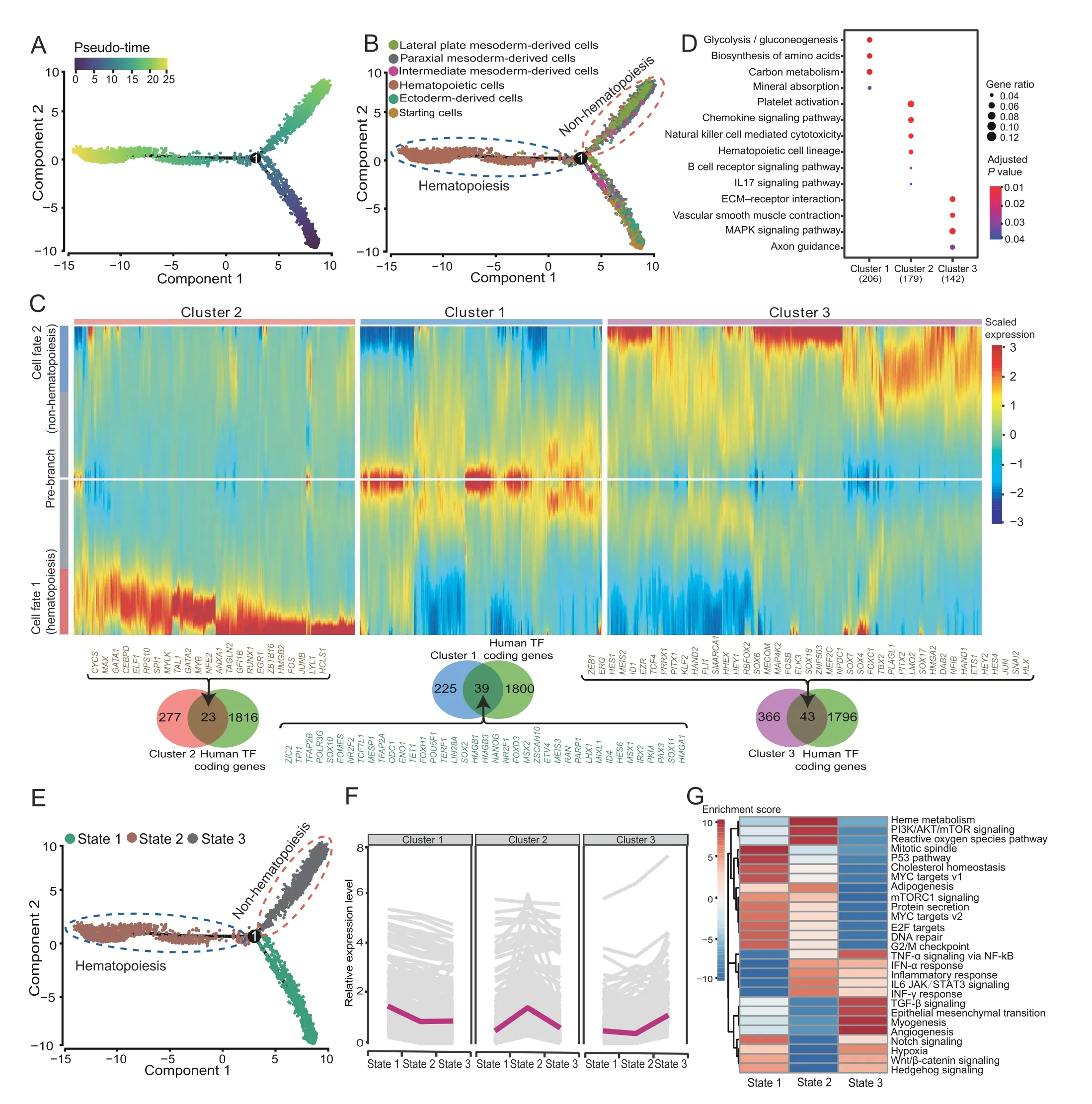
Figure 5 Cell pseudo-time trajectory of the iPSC-derived erythroid differentiation systemA. Developmental pseudo-time of iPSC-derived erythroid differentiation system by Monocle2. B. The distribution of different classification cells on the pseudo-time trajectory. C. Heatmap showing the specific gene expression patterns in hematopoietic and non-hematopoietic branches. D. Functional annotation analysis of each cluster gene from(C).E.The distribution of different state cells on the pseudo-time trajectory.F.The expression trend changes between states of each cluster gene from (D). G. Gene set variation analysis among different cell states.
To further understand the molecular characteristics of cells that entered the hematopoiesis branch,we analyzed the difference between state 2 initial cells (the first 100 cells that entered the hematopoiesis branch) and state 3 initial cells(the first 100 cells that entered the non-hematopoiesis branch), and found that compared to state 3 initial cells,state 2 initial cells had low expression ofCOL5A1,COL6A3, and other collagen genes, and high expression ofAIF1,SPI1, and other genes related to myeloid differentiation(Figure S9D).We then identified top 20 TFs(such as SPI1 and GATA1)regulating the highly expressed genes in state 2 initial cells (retrieved from the TRRUST database) (Figure S9E). Interestingly, these TFs and their target genes were significantly enriched in embryonic hemopoiesis, erythroid differentiation, and myeloid differentiation, suggesting that our system is similar to early embryonic hematopoiesis, which tends to produce erythroid and myeloid cells and is regulated by Notch signaling pathway and estrogen (Figure S9F).
The transition from starting cells to HSPCs
Previous studies have shown that HSPCs are differentiated from endothelial progenitor cells in the lateral mesoderm during embryonic development[17].To determine how the transition from starting cells to hematopoietic cells, we conducted the pseudo-time analysis of ESCs, iPSCs, endothelial cells, and CD34+HSPCs. Most of the iPSCs and ESCs fell into the pre-branch and underwent bifurcation.The cell fate 1 branch was mostly composed of CD34+HSPCs,and the cell fate 2 branch was mostly composed of endothelial cells (Figure 6A and B, Figure S10A).Differential gene expression analysis was performed between the two branches to determine the characteristic genes in each cell fate. Interestingly, we identified a hematopoietic-endothelial(HE)gene set(cluster 5),including 10 TF coding genes expressed on both endothelial cell and HSPC branches. The TF coding genesLMO2andFLI1in this gene set are the marker genes of HECs during embryonic development [70,71]. FOS, JUNB, and DAB2 are the downstream TFs of TGF-β, and TGF-β drives HE programming[72,73].FOS and MAP4K2 are the effect factors in the MAPK cascade where VEGF can directly regulate the establishment of arterial specifications of endothelial progenitor cells[74].The HE gene set(cluster 5)was enriched in cell adhesion molecules and tight junctions(Figure 6C).HSPC branch corresponds to state 2 cells,endothelial cell branch corresponds to state 3 cells, and HE gene set was highly expressed in state 2 and state 3 cells (Figure S10B and C). At the same time, we found that some CD34+HSPCs fell in state 3, which was defined as HECs. By comparing with CD34+HSPCs in state 2, we found that HECs highly expressedKDR,SOX17,and other endothelial characteristic genes,and these highly expressed genes were enriched in cell adhesion molecules and regulated by Ras,Notch, and VEGF (Figure 6D and E). We verified the expression of some cell adhesion molecules by qPCR assay combined with previous function annotation results for cell classification marker genes and cell–cell contact pattern analysis. Results indicated that the expression of these molecules fluctuated during RBC regeneration; their expression was up-regulated during the HE formation [75](EB day 4), while their expression was down-regulated in hematopoietic cells (SC day 14)(Figure S10D).
The transition from HSPCs to erythroid cells
To clarify the transition from HSPCs to erythroid cells,we conducted a pseudo-time analysis of hematopoietic cells.Hematopoietic cells consisted of HSPCs (CD34+HSPCs,13.5%),erythroid cells(BFU-E cells,42.8%;CFU-E cells,11.7%; erythrocytes, 3.1%; Ortho-E cells, 1.3%), myeloid cells (monocytes, 13.3%; pro-myelocytes, 5.3%; macrophages, 3.6%; neutrophils, 2.9%; myelocytes, 1.4%; eosinophils,0.3%),and a small amount of lymphocytes(CD8+T cells, 0.7%) (Figure S11A and B). The differentiation trajectory showed fewer cells on the pre-branch, and most of the cells were ordered to the erythropoiesis and non-erythropoiesis branches (Figure 7A and B). Specifically, some BFU-E cells, CFU-E cells, and CD34+HSPCs were distributed in the pre-branch corresponding to state 4.Non-erythroid cells(eosinophils,macrophages,monocytes,myelocytes, neutrophils, and pro-myelocytes) were mostly distributed in the non-erythropoiesis branch (cell fate 1)corresponding to state 3, and erythroid cells (BFU-E cells,CFU-E cells, erythrocytes, and ortho-E cells) were mostly distributed in the erythropoiesis branch (cell fate 2) corresponding to states 1, 2, and 5 (Figure 7C, Figure S11C).Simultaneously,state 4 cells highly expressedMYB,CD34,CD45/PTPRC, andCD44, which are yolk sac-derived myeloid-biased progenitor (YSMP) markers. YSMPs are human yolk sac second wave hematopoietic progenitor cells. And state 4 cells almost did not expressHOX6(the HSPC-specific marker in the definitive hematopoietic AGM region [76]), andKIT(the mouse yolk sac second wave hematopoietic progenitor marker [77]) (Figure 7D),suggesting that our system is similar to the second wave of hematopoiesis in the human yolk sac.
To further clarify the molecular regulation mode in the differentiation process of YSMP in the system,we performed a difference map between the two branches (erythropoiesis and non-erythropoiesis). YSMPs (pre-branch) were driven by TAL1, GATA1, and 12 other TFs to enter the erythropoiesis branch (cell fate 2); oppositely, YSMPs were driven by CEBPD,MAF,and 22 other TFs to enter the non-erythropoiesis branch (cell fate 1) (Figure 7E, Figure S11D). We identified 336 signature genes (cluster 2) in YSMPs (pre-branch), which were mainly enriched in oxidative phosphorylation. Interestingly, this gene set has a strong tendency for expression in the erythropoiesis branch,which may be erythropoietin (EPO) driven in the system(Figure 7E, Figure S11E). Besides, we found that some of the erythroid cells will fall into the non-erythropoiesis branch, indicating production of other types of erythroid cells in our system (Figure S11C).
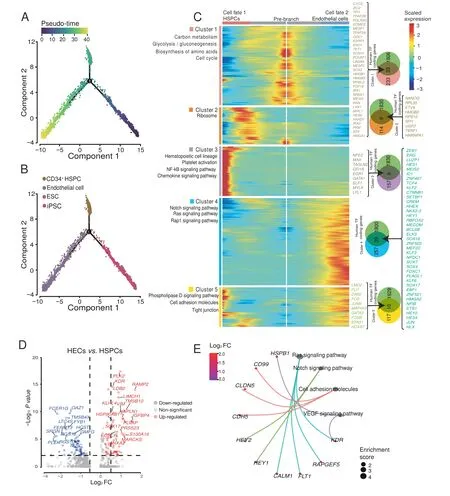
Figure 6 Key factors driving hematopoiesisA. Developmental pseudo-time of iPSCs, ESCs, endothelial cells, and CD34+ HSPCs by Monocle2. B. The distribution of different type cells on the pseudo-time trajectory.C.Heatmap showing the specific gene expression patterns in endothelial cell and HSPC branches.D.Volcanic plot of DEGs from gene differential expression analysis between HECs and HSPCs(P<0.05,|log2 FC|>0.5).E.KEGG analysis of highly expressed genes in HECs.HEC,hematopoietic-endothelial cell.
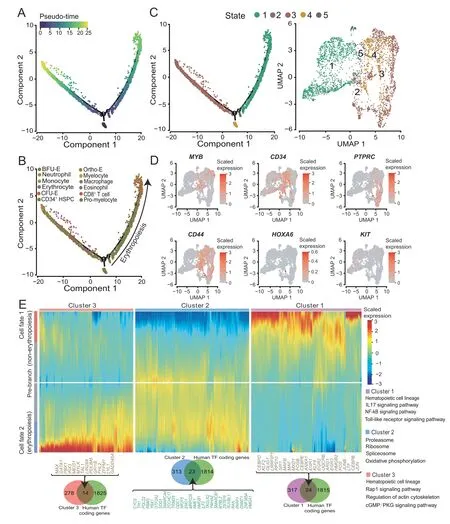
Figure 7 Differentiation trajectory of hematopoietic cellsA.Developmental pseudo-time of hematopoietic cells by Monocle2.B.The distribution of different type cells on the hematopoietic pseudo-time trajectory.C.The distribution of different state cells on the pseudo-time trajectory(left)and UMAP plot showing the distribution of hematopoietic cell states(right).D.UMAP plots of every single cell showing the expression levels of MYB,CD34,PTPRC,CD44,HOXA6,and KIT.E.Heatmap showing the specific gene expression patterns in erythropoiesis and non-erythropoiesis branches.
Differentiation trajectory of erythroid cells
To accurately understand the molecular activities during erythroid differentiation, we took BFU-E cells, CFU-E cells, erythrocytes, and ortho-E cells to perform a pseudotime analysis (Figure 8A). The pseudo-time result was consistent with the SingleR cell annotation result: the erythroid progenitor cells were mostly distributed at the beginning of the differentiation trajectory, and the differentiated terminal cells were mainly at the end of the trajectory(Figure 8B,Figure S12A).Furthermore,clustering analysis of the top 1000 genes with the pseudo-time scoring significance was performed to obtain the highly expressed gene set (cluster 1) of erythroid progenitor cells, including theGATA1and 36 other TF coding genes, which were mainly enriched in the estrogen signaling pathway. The expression of genes in cluster 1 was mostly initially high and then decreased. The gene set cluster 2, which was highly expressed in the erythroid cells, includes 34 TF coding genes, such asBCL11A, and genes in this cluster were mainly enriched in cholesterol metabolism, glycosaminoglycan binding,and oxygen carrier activity(Figure 8C,Figure S12B and C).The pseudo-time analysis divides cells into three states;the erythroid progenitor cells correspond to state 1 in which the cluster 1 gene set was highly expressed,and the erythroid cells correspond to states 2 and 3 in which the cluster 2 gene set was highly expressed (Figure 8D,Figure S12D). Specifically, state 1 (erythroid progenitor cells) responds more strongly to estrogen (Figure 8E). We tested the role of estrogen by adding estradiol during the induction and differentiation process of SCs and observed that expression ofKLF1slightly increased in the erythroid progenitor cells,but the expression ofALAS2,GATA1,andKLF1in the terminal erythroid differentiation was significantly inhibited(Figure S12E).Altogether,it suggested that estrogen promotes the proliferation of erythroid progenitor cells, but may inhibit erythroid differentiation(Figure S12E).
Interestingly,we found that state 2 cells highly expressedCD74and lowly expressedEPOR. Compared with state 3 cells,state 2 cells lowly expressed genes related to oxygen carrier activity and oxygen binding(e.g.,HBDandHBE1),and highly expressed genes related to MHC protein complex binding (e.g.,CD81,C1QBP, and other myeloid characteristic genes), which can be defined as immune erythroid cells(Figure 8G,Figure S12F).Also,we performed a gene differential expression analysis between ortho-E and other erythroid cells (Figure S12G). Ortho-E cells lowly expressed genes related to the ribosome (e.g.,RPS16andRPS13), oxidative phosphorylation, and cell cycle, and in contrast,highly expressed genes associated with PI3K-Akt and FoxO signaling pathways,among whichFOXO3plays a crucial role in the enucleation of erythroid cells [78](Figure 8H, Figure S12G and H).
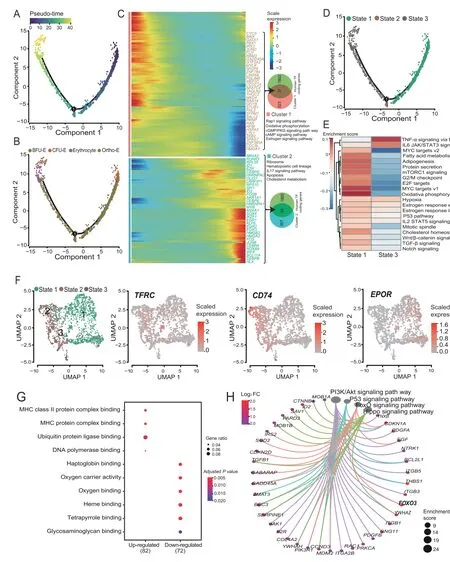
Figure 8 Pseudo-time trajectory of erythroid cellsA. Developmental process of erythroid cells by Monocle2. B. The distribution of different type cells on the erythroid cell pseudo-time trajectory. C.Heatmap showing the gene expression dynamics during erythroid differentiation.D.The distribution of different state cells on the erythroid cell pseudotime trajectory.E.Gene set variation analysis between cells from state 1 and state 3.F.UMAP plots of erythroid cell state distribution and the expression levels of TFRC,CD74,and EPOR.G.GO analysis of DEGs between cells from state 1 and state 3.H.KEGG network constructed with highly expressed genes in ortho-E cells compared to other erythroid cells.
Discussion
With the rapid development of single-cell technology, single-cell transcriptome studies on the development of the hematopoietic lineage have been extensively reported, and the known hematopoietic theory has been revised and supplemented, but most studies focused on bone marrow,peripheral blood, cord blood, and other systems [79,80].Here, we used the EB culture method verified by many laboratories to construct the RBC regeneration system derived from iPSCs[8,44,45,81].The erythroid differentiation efficiency of iPSCs generated from different types of tissues might be different;however,these iPSC lines can differentiate toward erythroid cells under appropriate inducing conditions [82]. After multiple induction-differentiation experiments under the same conditions, it is clear that HSPCs in the SCs produced by the EBs can stably enter the erythroid differentiation process(data not shown).After clarifying the repeatability and stability of the system, we performed a single-cell transcriptomics study on the erythroid differentiation system derived from iPSCs, in which over 10,000 high-quality cells of the differentiation system from two batches were analyzed. The two batches of cells are positively correlated, and the mesoderm-derived and hematopoietic cells are dominantly present in each batch.To avoid the in-between cell types in the differentiation system, we applied four datasets that are integrated by the R package SingleR, including Human Primary Cell Atlas reference dataset, BLUEPRINT and Encode reference dataset, Database of Immune Cell Expression reference dataset, and Novershtern hematopoietic reference dataset,and two public erythroid cell reference datasets,to carefully characterize the cell types in the iPSC-derived erythroid differentiation system. The cell map and cell differentiation trajectory of the system were mapped for the first time, and the whole transcriptome dynamics from iPSCs to the erythroid lineage was delineated.
This study observed that iPSCs went through two key transformations during EB formation to hematopoiesis and erythropoiesis.The first transformation was EHT,in which iPSCs differentiated into the germ layers under the induction of BMP4 and FGF-2 to form endothelial cells in the lateral plate mesoderm. Endothelial cells had strong cell–cell contacts between themselves and other cells through the ESAM during EHT. Under the induction of VEGF, HECs were formed with the high expression ofFLI,LMO2, and the cell adhesion molecules, and HECs were converted to HSPCs with the up-regulated expression ofGATA2under the influence ofRUNX1and the down-regulated expression of cell adhesion molecules. Reports showed that cell adhesion plays an essential regulatory function in mammalian embryonic development.Furthermore,cell–cell interactions were extremely indispensable in the formation and function of erythroblastic islands,the niches where macrophages and immature erythrocytes were gathered and contacted to help erythrocyte maturation and enucleation during erythropoiesis[83–85].
The second transformation was HSPC differentiation into erythroid cells. The HSPCs produced in the system were similar to CD34+/CD45+/CD44+YSMPs [86], and they tended to differentiate into erythroid cells under the induction of EPO. YSMPs entered the erythroid differentiation pathway driven by GATA1. During this period, the expression of estrogen target genes was up-regulated at the beginning and then decreased.A recent study revealed that estrogen could promote self-renewal of HSCs, expand splenic HSCs, and promote erythropoiesis during pregnancy [87]. A small amount of immune CD74+erythroid cells were produced.Immune erythrocytes and their surface markers have been discovered and identified in recent years[88,89].This study found for the first time that thein vitroerythroid differentiation system can also produce such cells,which provides a new direction for the clinical availability of RBCs regeneratedin vitro. Also, we found that TFDP1 may be related to the restriction of cell number during the maturation of erythroid cells[65].Moreover,we found thatFOXO3was highly expressed in ortho-E cells, which has been reported to play a vital role in the denucleation of erythroid cells[78],and the high expression ofUPK1A-AS1andTRAPPC3Lin ortho-E cells may also regulate RBC denucleation-related genes.
Our results show that iPSCs go through two essential transformations to form erythroid cells in our system, accompanied by a complex TF network and the dynamic expression of cell adhesion molecules and estrogenresponsive genes.It suggests that we can add cell adhesion activators before entering the first transformation, but add cell adhesion inhibitors and inhibit endothelial TFs (e.g.,SOX7) from increasing the production of HSPCs in the system during the first transformation. Subsequently, we could activate erythroid TFs (e.g., GATA1) and add estradiol during the second transformation to increase the production of erythroid cells. This study provides a powerful theoretical guide for optimizing our iPSC-derived erythroid differentiation system.
In summary, using the entire iPSC-derived erythroid differentiation system in which both EBCs and SCs are included, we systematically characterize the molecular characteristics and differentiation trajectory of various cell types in the system. We provide essential factors driving iPSC differentiation into erythroid cells. However, technically it is not enough to improve thein vitroiPSC-derived erythroid differentiation system at only one stage, and the erythroid differentiation system needs to utilize different iPSC lines to prepare regenerative RBCs of various blood types to suffice the needs of clinical blood transfusion under different blood types or different disease conditions in the future.Deciphering the erythroid differentiation systems at different stages started from SCs in the EB culture system,we might identify more useful factors and pathways regulating adult globin expression and denucleation, which are the bottlenecks in this field.Moreover,we might characterizethe real key regulators controlling the RBC production process by comparing thein vitroiPSC-derived erythroid differentiation system under physiological conditions (e.g., bone marrow). With the development of single-cell sequencing technology, we will be able to excavate more potential mechanisms regulating RBC development and maturation and obtain more mature regenerated functional RBCsin vitrofor clinical applications in the future.
Materials and methods
iPSC and EB culture and erythropoiesis
iPSCs(sourced from Linzhao Cheng’s lab)were cultured in vitronectin (Catalog No. A14700, Invitrogen, Carlsbad,CA)-coated culture dishes using essential 8 medium(Catalog No. A1517001, Gibco, Carlsbad, CA). When the cells reached 70%–80% confluence (usually around 3–4 days),they were digested to SCs using 0.5 mM EDTA (Catalog No.15575020,Invitrogen)and passaged with a ratio of 1:4.
EB culture was performed from day 0 to day 11, as previously described [8]. From day 11 to day 14 of the EB culture,thrombopoietin(TPO)was replaced by 3 U/ml EPO(Catalog No.100-64,PeproTech,Cranbury,NJ).On day 14,SCs were collected using a 70-μm cell strainer. Subsequently,the SC solution was centrifuged at 300gfor 5 min to remove the supernatant, and SFM containing 50 μg/ml stem cell factor (SCF; catalog No. 300-07, PeproTech),100 μg/ml IL3 (Catalog No. 200-03, PeproTech), 2 U/ml EPO, and 1% penicillin/streptomycin (P/S) (Catalog No.15140122, Life Technologies, Carlsbad, CA) was used to resuspend the SC precipitates.The cells were cultured in a 6-well plate at a total volume of 2 ml per well with a final concentration of 5×105cells/ml.All cells were cultured for 6 days at 37°C under 5% CO2, and the medium was changed every three days. On day 20, the medium was replaced with SFM containing 2 U/ml EPO and 1%P/S,and the cell concentration was adjusted appropriately. The number of cells in each well did not exceed 2×106,and the total volume of the medium per well was 2 ml. The cells were cultured at 37°C under 5%CO2,and the medium was changed every three days.
CFU assay
Incubation of day 14 SCs was performed using MethoCult™H4434 classic methylcellulose medium for human cells(Catalog No. 04434, StemCell, Vancouver, Canada). Each well of a 12-well plate contained around 10,000 cells and 0.6 ml of the medium. Other operations were performed as described in the instructions. Cells were incubated for 12−14 days,and the clones were observed under the microscope.
Flow cytometry analysis
First, approximately 1 × 105cells were collected from the culture, centrifuged for 5 min at 300gto remove the supernatant, and washed twice with 2 ml 1× Dulbecco’s phosphate-buffered saline (DPBS; catalog No. 14190136,Gibco)solution containing 2%fetal bovine serum(Catalog No. 16000044, Gibco) and 2 mM EDTA. After centrifugation,the cells were resuspended in 50 μl of 1×DPBS buffer,and the antibodies were added, followed by incubation at 4°C for 10 min in the dark.The cells were washed twice by adding 2 ml of 1× DPBS buffer after incubation and then resuspended in 200 μl of 1× DPBS buffer to prepare a cell suspension for the assay. The BD FACSAria II instrument was used for flow cytometry analysis,and the data analysis was performed with Flowjo software (v.7.6, Three Star).The antibodies used in this study included anti-human CD34-FITC (Catalog No. 130-098-142, Miltenyi Biotec,Bergisch Gladbach, Germany), anti-human CD45-PE(Catalog No. 170-081-061, Miltenyi Biotec), anti-human CD71-PE(Catalog No.130-099-219,Miltenyi Biotec),and anti-human CD235a-APC (Catalog No. 130-118-493, Miltenyi Biotec).
Wright-Giemsa staining
Approximately 1 × 105cells were taken, and the medium was removed by centrifugation at 300gfor 5 min;the cells were resuspended in 20 μl of 1× DPBS buffer. The cells were placed onto one clean slide, and another clean slide was used to push the first to 30°−45° to spread the cells evenly.The pushed film was placed in a 37°C incubator for 1−3 h. Then, 200 μl of Wright-Giemsa staining A solution(Catalog No.BA-4017,BASO,Zhuhai,China)was placed onto the cell-containing slide, and the slide was placed at room temperature for 1 min. It was immediately covered with 400 μl of Wright-Giemsa staining B solution. After incubated at room temperature for 8 min, the slide was gently rinsed with fluid distilled water along the area around the slide without cells.The slide was rinsed for an additional 1 min until the water ran clear and transparent. Then, the slide was placed at room temperature until dry, and a microscope was used to observe and record cell morphology.
Total RNA extraction and qPCR
Total RNA was extracted from the iPSC line BC1,EBCs on day 4, SCs on day 14, SCs on day 17, and SCs on day 23,respectively. The total RNA was then reverse transcribed into cDNA using the PrimeScript RT Reagent Kit with gDNA Eraser (Catalog No. RR047A, TaKaRa, Kusatsu,Japan).The diluted cDNAwas used as a template for qPCR.The instrument used for qPCR was the Bio-Red CFX96 Real-Time PCR detection system.All primers used in qPCR are listed in Table S2.
scRNA-seq data analysis and bulk RNA-seq data analysis
Day 14 EBs were dissociated with Accutase solution(Catalog No. A6964, Sigma, St. Louis, MI). Single-cell suspensions at 800 cells/μl were subjected to Chromium 10X Genomics library construction. Raw gene expression matrices generated by CellRanger (v.3.0.0) were imported into Seurat(v.3.2.2)[46].After removing low-quality cells and genes(percentage of mitochondria reads ≥25%,unique genes ≤200 in a cell, and genes expressed in less than 5 cells), the remaining cells were annotated by SingleR and classified according to annotation results[48].TF regulatory network was constructed by SCENIC (v.1.1.2) [63], and cell–cell contact patterns were constructed by CellChat(v.0.0.2) [60]. Particular clusters were imported into Monocle2[66]for pseudo-time ordering.For bulk RNA-seq data, FASTQC and Trimmomatic were used to remove adaptor sequences and low-quality reads,respectively.Then,clean data were mapped to the reference genome(GRCh38)by HISAT2 to generate a gene expression matrix,which was then imported into R Package DEGseq to identify differentially expressed genes (DEGs). Heatmap analysis and principal component analysis (PCA) were performed by pheatmap and PRINCOMP,respectively.We used clusterProfiler(v.3.18.0)to perform functional annotation analysis[59].All data analyses were completed in R (v4.0.0) environment.The list of human TFs was downloaded from http://humantfs.ccbr.utoronto.ca/download.php. The Circular network was visualized by GeneMANIA[90].
Data availability
The scRNA-seq data of day 14 EBCs and SCs and the bulk RNA-seq data of iPSC line BC1 and day 14 SCs are available in the Genome Sequence Archive [91] at the National Genomics Data Center, Beijing Institute of Genomics,Chinese Academy of Sciences/China National Center for Bioinformation (GSA: CRA002066), and are publicly accessible at http://bigd.big.ac.cn/gsa/.
CRediT author statement
Zijuan Xin:Investigation,Data curation,Formal analysis,Validation,Visualization,Writing-original draft,Writingreview&editing.Wei Zhang:Investigation,Data curation,Formal analysis, Validation, Visualization, Writing - original draft, Writing - review & editing.Shangjin Gong:Formal analysis.Junwei Zhu:Validation.Yanming Li:
Project administration.Zhaojun Zhang:Conceptualization,Resources,Writing-original draft,Writing-review&editing,Supervision.Xiangdong Fang:Conceptualization,Resources, Writing - original draft, Writing - review &editing, Supervision. All authors read and approved the final manuscript.
Competing interests
The authors have declared no competing interests.
Acknowledgments
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No.XDA16010602),the National Key R&D Program of China(Grant Nos. 2016YFC0901700, 2017YFC0907400, and 2018YFC0910700), and the National Natural Science Foundation of China (Grant Nos. 81670109, 81870097,81700097, 81700116, and 82070114). We were grateful to Linzhao Cheng for agreeing to use iPSC line BC1 and Qianfei Wang for providing the iPSCs.We thank the editor Suzanne N in enago editing service who provided assistance for the manuscript.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2021.03.009.
ORCID
0000-0001-9418-4848 (Zijuan Xin)
0000-0002-0519-3935 (Wei Zhang)
0000-0002-9811-5302 (Shangjin Gong)
0000-0001-9766-3334 (Junwei Zhu)
0000-0002-8213-9166 (Yanming Li)
0000-0003-0490-6507 (Zhaojun Zhang)
0000-0002-6628-8620 (Xiangdong Fang)
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- scDPN for High-throughput Single-cell CNV Detection to Uncover Clonal Evolution During HCC Recurrence
- Single-cell Long Non-coding RNA Landscape of T Cells in Human Cancer Immunity
- Single-cell Transcriptomes Reveal Characteristics of MicroRNAs in Gene Expression Noise Reduction
- Single-cell RNA Sequencing Reveals Sexually Dimorphic Transcriptome and Type 2 Diabetes Genes in Mouse Islet β Cells
- Single-cell RNA Sequencing Reveals Thoracolumbar Vertebra Heterogeneity and Rib-genesis in Pigs
- GranatumX: A Community-engaging, Modularized, and Flexible Webtool for Single-cell Data Analysis