SiC陶瓷与Zn界面结合特性的第一性原理研究
2021-02-22马志鹏夏杨嘉雯李昊宣张茗瑄许志武于心泷
马志鹏, 夏杨嘉雯,李昊宣,张茗瑄,许志武,于心泷
(1.东北石油大学 材料科学与工程,黑龙江 大庆 163318;2.先进焊接与连接国家重点实验室(哈尔滨工业大学),哈尔滨 150001)
SiC陶瓷具有高硬度,高熔点,耐磨性好,高温时抗氧化性强、导热系数高和热膨胀系数小等特点,因此在航空航天、核能、机械、光学及电子等领域得到广泛应用[1-2]。但是SiC陶瓷材料延展性和韧性差,加工性能差,使得制造大尺寸且形状复杂的构件十分困难,因而常常需要采用钎焊技术来实现SiC陶瓷的连接[3-4]。
目前SiC陶瓷钎焊连接主要集中于高温连接领域[5],钎焊温度高,周期长,且由于SiC陶瓷与金属的线膨胀系数和弹性模量存在较大差距,会造成接头内应力分布不均匀,导致接头性能下降。在电子封装领域中,为避免高温连接造成元件损伤,SiC陶瓷需在低温下焊接。金属锌的熔点为420 ℃,锌基钎料属于低温钎料,采用该钎料一定程度上缓解了接头中的残余应力问题[6]。
Urena等[7]研究了Zn-3Al、Zn-4.73Al-0.62Cu和Zn-3.9Al-0.89Cu三种软钎料与SiC的润湿性,结果发现钎料的熔点越高焊接接头的润湿性越好。Wu等[8]在超声波的作用下采用Zn-5Al钎料焊接SiC陶瓷,当超声作用较短时,接头界面处形成了η-Zn相和大量脆性层状共晶相,接头剪切强度为102 MPa,随着超声波作用时间增加,超声波消除了共晶相,且使界面处晶粒细化,接头剪切强度升高。Zhang等[9]采用超声波钎焊的方法,以Zn-Al为液态钎料焊接SiC陶瓷,在超声波的作用下,界面氧化层消失,部分SiC颗粒与钎料融合,使钎料与母材形成良好的润湿结合,接头剪切强度随超声作用时间延长而增加。Ji等[10]采用Zn14Al过共晶钎料对Al2O3陶瓷和Cu进行了超声波钎焊,Al2O3侧界面出现了晶体Al2O3,Cu侧界面在超声作用下出现了明显的空蚀坑,钎焊完成后接头的最高强度可达66 MPa。Xu等[11]采用超声波钎焊的方法,使用Zn-Al钎料焊接Al2O3/6061Al复合材料,随着超声振幅的增加界面处氧化膜逐渐消失,Al2O3颗粒与Zn-Al合金形成良好的结合,且Al的含量越多焊接接头的剪切强度越高。陈碧强等[12]采用3种Zn基钎料钎焊SiC颗粒增强铝基复合材料,发现Zn可以扩散到母材基体内,改善了接头的润湿性,分析了Mg、Ga元素对焊接接头性能的作用,在Zn基钎料中的Mg元素对陶瓷/金属钎料界面的润湿性和析出物有显著影响,为优化元素与钎焊规范指明方向。
上述的定性实验研究对界面微观作用机制的分析略显粗糙,由于界面结合涉及大量原子与界面之间复杂的相互作用,所以没有实现对金属/陶瓷界面结合本质的把握[13]。因此本文采用基于从头算理论的第一性原理模拟计算方法,搭建6种Zn/SiC的界面模型,通过分析表面能、界面能、电子结构以及Mulliken布局数,从微观的角度全面认识Zn/SiC界面结合特性及规律的本质。
1 计算方法与模型
本文采用基于密度泛函理论(DFT)的第一性原理方法,运用Cambridge Serial Total Energy Package(CASTEP)模块,对界面模型进行结构优化与计算。计算时采用广义梯度近似(GGA)中的PBE为交换关联泛函,使用自洽场方法(SCF)求解Kohn-Sham方程,其中SCF能量的收敛值为5.0×10-7eV/atom。BFGS收敛容差设置为:体系总能量误差在5.0×10-6eV/atom以内,应力偏差小于0.05 GPa,原子力在0.3 eV/nm 以下,公差偏移小于10-4nm。经过收敛性测试,平面波截断能Ecut设置为450 eV,Zn的K值取7×7×2,SiC的K值取9×9×2。
SiC属于密排六方晶型,空间群为P63MC,晶格常数a=b=0.307 8 nm,c=1.004 6 nm,堆垛方式为ABCB。Zn属于变态密排六方晶格,空间群为P63/MMC,每个Zn原子都有12个邻近的原子,晶格常数a=b=0.266 49 nm,c=0.494 68 nm[14]。图1为SiC和Zn的晶体结构。结构优化后得到SiC的理论晶格参数为a=b=0.308 3 nm,c=1.004 6 nm,且SiC的内聚能Ecoh=15.17 eV,与Thibault计算的a=b=0.305 3 nm,c=0.999 4 nm,Ecoh=15.11 eV[15]相差不大,因此本文的计算参数是可靠的。此外,为了减小极性的影响,在构建表面模型时将真空层厚度设置为2 nm。

图1 SiC和Zn晶体结构
2 结果与讨论
2.1 Zn和SiC表面能与稳定性
在构建两相界面时,采用表面能低的表面作为结合界面,此时界面更稳定。对SiC和Zn模型的(0001)、(0100)、(0101)和(1101)晶面的表面模型进行结构优化后计算表面能。
(1)
式中:Eslab为在切表面添加真空层的体系的总能;Ebulk为晶胞体系的总能;Nslab和Nbulk分别为两种体系包括的原子数;A为表面模型的表面积。
表1是SiC各晶体表面的表面能,表2为Zn各表面的表面能。

表1 SiC各晶体表面的表面能

表2 Zn各晶体表面的表面能
由计算结果可知,SiC(0001)表面模型的表面能最低,表面能为Esurf=0.024 J/m2;Zn(0001)表面模型的表面能最低,表面能为Esurf=0.001 J/m2,选择SiC的(0001)和Zn的(0001)为结合界面。
在构建界面模型之前,需对界面两侧结构的原子厚度进行收敛性测试,以选择合适原子层数的表面模型作为界面结合模型。SiC及Zn各原子层数的表面能计算结果如表3和表4所示。可知SiC的原子层数在12层时,表面能收敛于3.72 J/m2;Zn的原子层数在9层时,表面能收敛于0.04 J/m2。选择12层SiC(0001)表面模型和9层Zn(0001)表面模型均可代表相应的体相结构,可用于构建Zn(0001)/SiC(0001)界面模型。
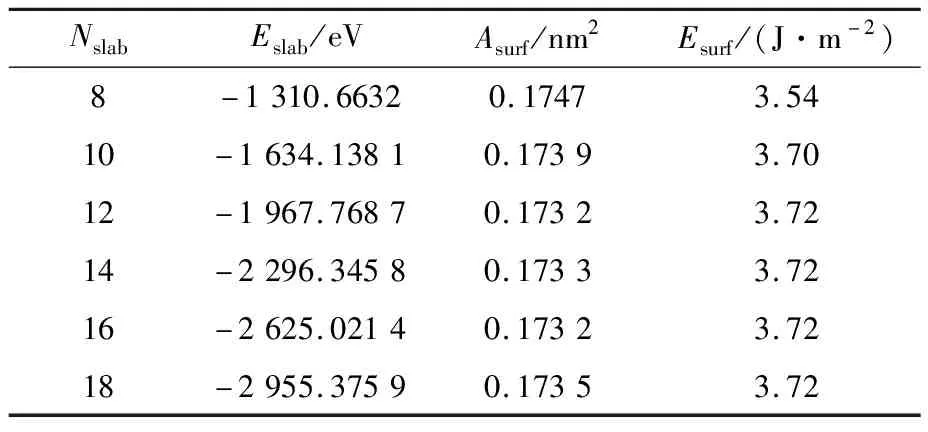
表3 SiC的原子层数和表面能

表4 Zn的原子层数和表面能
2.2 Zn/SiC分离功与界面稳定性
根据收敛性测试结果,建立Zn(0001)/SiC(0001)界面模型如下:将9层的Zn(0001)堆垛在12层的SiC(0001)表面,且在上下表面添加2 nm的真空层,该界面的失配度为1.55%,通常失配度小于5%,认为形成了典型的共格界面,可以构成稳定界面。考虑到SiC(0001)表面有两种不同的终端(Si终端和C终端)和3种不同的堆垛方式(孔穴型、顶位型和中心型),共建立了6种界面模型。图2为Zn/SiC的C终端界面模型,图3为Zn/SiC的Si终端界面模型。

图2 Zn/SiC的C终端界面模型

图3 Zn/SiC的Si终端界面模型
本文用界面分离功来衡量界面的结合强度,分离功是将一个界面分离成为两个自由表面时所需要的能量[16],而具有合适界面间距的界面模型是最稳定的。采用Universal Binding Energy Relation(UBER)方法,即分别给6种界面模型选取一系列的界面间距d0(通常为0.15~0.6 nm),对每一个界面模型进行优化并计算各间距下界面的分离功。
分离功计算公式为
(2)
式中:Eab为由表面a和表面b形成的界面总能量,eV;Ea为表面a的总能量,eV;Eb为表面b的总能量,eV;S为界面面积,nm2。
以d0为自变量,Wsep为因变量,得到UBER曲线,如图4(a)和(b)所示。
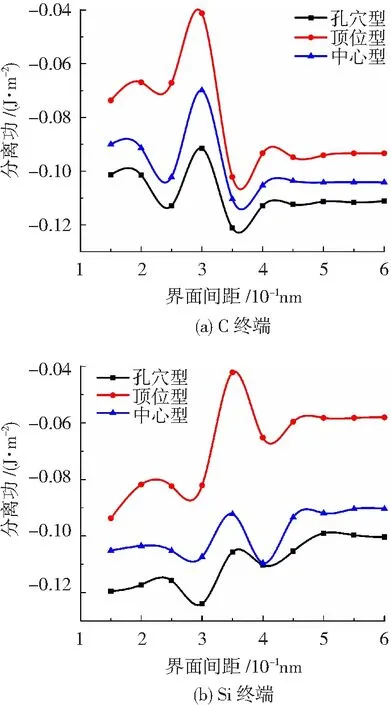
图4 两种终端界面的界面间距和分离功
可知,不同堆垛方式对界面平衡间距和分离功有影响。在相同的终端界面系统中,3种界面模型UBER曲线的规律大致相同,其中孔穴型的分离功最大值比顶位型和中心型都大,说明孔穴型是3种堆垛方式界面模型中最稳定的。对于不同终端的孔穴型界面模型,Si终端分离功最大值比C终端大,且Si终端界面平衡间距小于C终端,这表明Si终端孔穴型界面稳定性更好。C终端孔穴型中最稳定界面模型的界面间距d0=0.351 nm,分离功Wsep=0.124 J/m2;Si终端孔穴型中最稳定界面模型的界面间距d0=0.289 nm,分离功Wsep=0.126 J/m2。以上两种界面模型是所有界面模型中最稳定的,具有最大结合强度。
2.3 电子结构
Zn/SiC界面的稳定性、力学性能与界面处的成键特征紧密相关,本文计算了上述两种终端里最稳定的两个界面的电子结构,包括电荷密度图、电荷密度差分图、电荷布局和键布局。
电荷密度图代表了电荷的聚集程度,界面处的电荷密度越大,各原子之间相互作用越强。图5(a)为C终端模型的电荷密度图(虚线代表界面),界面1上的Zn(5)与C(5)或Si(1)之间都没有明显的电子云重叠,故未成键。界面2上的Si(5)与Zn(1)之间有明显的电子云重叠,由此推断形成了Zn(1)-Si(5)键,而C(2)与Zn(1)之间未成键。图5(b)为Si终端模型的电荷密度图(虚线代表界面),同理可知,在界面3上只形成了Zn(5)-Si(2)键,界面4上未成键。
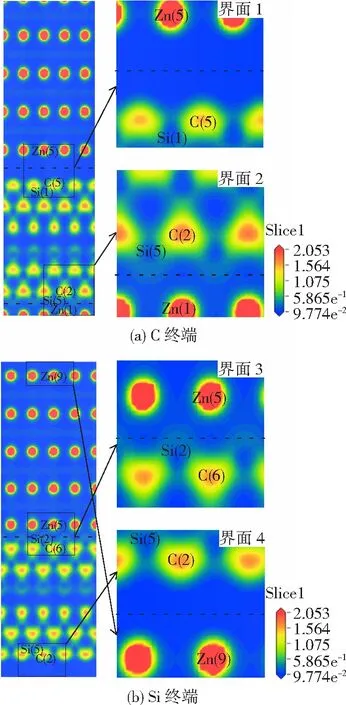
图5 两种终端界面的电荷密度图
对比界面2和界面3两图,可明显看出Si终端的Zn(5)和Si(2)之间的电荷密度大于C终端的Zn(1)和Si(5)之间的电荷密度,电子云重叠程度更大,颜色更深,这说明Si终端的Zn(5)-Si(2)键结合强度强于C终端的Zn(1)-Si(5)键。两种终端界面结合主要由Zn原子和Si原子之间的相互作用贡献。
电荷密度差分图不同于电荷密度图,它表示电荷的转移情况,图6为C终端和Si终端的电荷密度差分图(图中虚线代表界面),图中蓝色代表缺失电子,白色代表不变,红色代表富集电子。

图6 两种终端的电荷密度差分图
在C终端中,界面1上的Zn(5)与C(5)或Si(1)之间呈白色并且没有电子云重叠,这表明Zn(5)与其他两种原子之间没有电荷转移和电子共用现象,未成键。界面2上Zn(1)周围呈白色,Si(5)周围呈蓝色,原子之间呈白色,有电子云重叠,表明二者之间成键,且没有电子共用的现象,即没有体现共价键的特性。C(2)周围富集大量电子,Zn(1)周围缺失电子,然而这两个原子之间呈蓝白色,并没有大量电子转移现象,故未成键。在Si终端中,同理可知,界面3中的Zn(5)和Si(2)之间没有形成共价键,C(6)和Zn(5)未成键,界面4中也未成键。结合表5中主要原子的Mulliken电荷布局,C终端模型中Zn(1)得到0.01个价电子,Si(5)失去0.90个价电子;Si终端模型中Zn(5)得到0.03个价电子,Si(2)失去了0.89个价电子,这证实了Zn原子周围虽然呈白色,但实际上Zn原子有微弱的电荷转移,Zn原子与Si原子之间形成了离子键。对比界面2和界面3的电荷密度差分图与两个终端电荷布局数可知Si终端中Zn(5)-Si(2)键的离子性大于C终端中Zn(1)-Si(5)键,即Si终端界面的结合强度大于C终端。以上分析结果与电荷密度图分析结果相符。

表5 主要原子的Mulliken电荷布局
键布局可以反应电子在界面处的分布情况,量化键合作用的类型和强度。通常认为键布局数越接近于0,键的离子性越强;反之,越偏离0,键的共价性越高;键长越短则键的强度越高。主要化学键的Mulliken键布局如表6所示,C终端Zn(1)-Si(5)键的布局数为0.28,键长为0.288 497 nm;Si终端Zn(5)-Si(2)键布局数为0.25,键长为0.285 703 nm。这表明两种终端都形成了离子键,其中Si终端Zn(5)-Si(2)键的离子性强,强度高。

表6 主要化学键的Mulliken键布局
3 结 论
1)由12层原子的SiC(0001)和9层原子的Zn(0001)建立的界面模型最稳定。
2)对于同种终端的界面,孔穴型界面模型都是最稳定的界面。不同终端的孔穴型界面模型,Si终端的稳定性强于C终端。
3)两种终端的Zn-Si键均为离子键,且Si终端的Zn-Si键的结合强度大于C终端。
4)Zn/SiC界面Zn-Si原子间形成的离子键在界面结合中占主要地位。
