分散固相萃取-超高效液相色谱-串联质谱法测定茶叶强极性农药残留
2021-02-21罗先锟
何 江,宋 磊,罗先锟
(1.贵阳市农产品质量安全监督检验测试中心,贵州 贵阳 5500811;2.贵州医科大学 公共卫生学院,贵州 贵阳 550026)
1 引言
茶叶中含有40多种无机矿物质和450多种有机化合物[1],具有提神清心、清热解暑、消食化痰等作用,还对心脑血管病有一定的药理功效,因此受到人们的喜爱。我国是世界上最大的茶叶生产国,茶园面积、产量及消费量均居世界第一[2]。同时,我国也是茶叶出口大国,茶叶出口为我国农民带来了经济收入[3]。长期以来,为了降低病虫害对茶叶的影响,保障茶叶的产量和品质,采用农药防治茶树病虫害成为茶园的一项重要措施[4]。常用极性农药中,如乐果可用于防治小绿叶蝉、茶蚜、介壳虫等害虫;氧乐果主要用于防治刺吸式口器的害虫;敌百虫主要用于防治茶毛虫、刺蛾、卷叶蛾等食叶性害虫;多菌灵可抑制茶树轮斑病的病原菌生长;涕灭威是一种高毒杀虫、杀螨、杀线虫剂。由于这些强极性农药杀虫杀菌效果好,能有效的防止茶树病害虫的生长,在茶叶生产中发挥着重要的作用。但是,强极性农药使用不当会造成茶叶中农药残留超标[5],影响茶叶的品质,损害消费者健康,还影响着茶叶产业的健康发展。如涕灭威及其代谢物过度使用,其致癌和致突变性可对环境和人体健康造成潜在危害[6,7];长期食用残留有多菌灵的食物,经消化道吸收后,能引起头昏脑胀、恶心呕吐等中毒症状,严重时可引起肝病和染色体畸变,同时对哺乳动物神经具有一定的毒性作用[8,9]。
茶叶农药残留的样品前处理技术呈现多方向发展、多个学科综合的新局面。常用的前处理方法有[10~13]:固相微萃取技术、固相萃取法、基质固相分散萃取技术,它们共同的优点是分离效率高、适用性广,但试剂的用量大,花费的成本高,步骤繁琐,耗时长。QuEChERS方法是近年来发展起来的一种样品前处理技术,该方法是先将均质的样品用乙腈提取,然后盐析分层,吸附剂[14](C18、GCB等)净化,比传统的净化方法更为快速简易[15]。常用检测方法包括生物检测技术、气相色谱法、气相色谱-质谱法[16]、液相色谱法、高效液相色谱串联质谱法等。UPLC-MS/MS是集UPLC的高分离能力与MS/MS的高灵敏度和高选择性于一体的强有力分离分析方法,对复杂基体中的农药残留具有很强的定性能力。本实验主要针对茶叶中强极性农药的检测,探索出更适合检测茶叶中强极性农药的色谱条件,采用QuEChERS前处理技术,结合UPLC-MS/MS法,建立一种更经济、快速、简便的检测茶叶12种强极性农药的方法。
2 实验部分
2.1 样品、试剂与仪器
茶叶样品均来自于市场和茶园抽样,按照GB 23200.13-2016 进行制样与保存。
乙腈、甲醇、甲酸(色谱纯,美国赛默飞世尔公司);N-丙基乙二胺(PSA)、十八烷基硅烷键合硅胶(C18)、石墨化炭黑(GCB)(上海安谱实验科技股份有限公司);无水硫酸镁(分析纯,天津市永大化学试剂有限公司);乙酸钠(分析纯,天津市申泰化学试剂有限公司);醋酸(优级纯,国药集团化学试剂有限公司);涕灭威、涕灭威亚砜、涕灭威砜、多菌灵、甲胺磷、噻虫嗪、敌百虫、氧乐果、乐果9种标准品溶液(1000μg/mL,北京坛墨公司);实验室用水为Milli-Q超纯水。
三重四级杆液相色谱质谱联用仪(Waters Xeve TQ,Waters公司);涡旋振荡器(SK-1,上海梅香仪器有限公司);高速离心机(HC-3515,安徽中科中佳科学仪器有限公司);电子天平(AX205,瑞士梅特勒托利多公司);Milli-Q超纯水净仪(美国Millipore公司)。
单标储备液:甲醇配制质量浓度为100 μg/mL的涕灭威、涕灭威亚砜、涕灭威砜、多菌灵、甲胺磷、噻虫嗪、敌百虫、氧乐果、乐果单标储备液,于 4 ℃保存。
2.2 样品前处理
称取2.00 g茶叶样品于50 mL离心管中,加入10 mL纯水,涡旋1 min,静置30 min;加入15 mL 1%醋酸-乙腈和一颗陶瓷质子于样品管中,涡旋1 min,加入6 g MgSO4、1.5 g乙酸钠,快速震荡后,涡旋2 min,10000 r/min离心5 min。
固相萃取法:取2 mL上清液过PRiME HLB小柱,收集滤液,UPLC-MS/MS测定。
QuEChERS法:取6 mL乙腈层加入10 mL离心管中,加入1.2 g MgSO4、400 mg PSA、400mgC18和200mgGCB,涡旋2 min,10000 r/min离心5 min。准确吸取2 mL上清液于10 mL试管中,40 ℃氮吹浓缩近干,加入1 mL初始流动相复溶,用0.22 μm滤膜过滤,UPLC-MS/MS测定。
2.3 UPLC-MS/MS 测定条件
色谱条件:ACQUITY UPLC HSS T3柱(1.8 μm,2.1×100 mm);柱温:30 ℃;进样体积:1 μL;流动相:A为水,B为乙腈。梯度洗脱程序:0~3 min,5% B;3~7 min,70% B;7~9 min,5% B;9~10 min,5% B;流速:0.3 mL/min;外标法定量。
质谱条件:电喷雾电离源(ESI);多反应监测(MRM);正离子模式;离子源温度:110 ℃;脱溶剂气温度:350 ℃;锥孔反吹气:50 L/Hr;脱溶剂气:1000 L/Hr。详细质谱参数见表1。
3 结果与讨论
3.1 色谱-质谱条件的选择
用甲醇配制浓度为0.2 μg/mL的混合标准溶液,选用保留特性差异大的3种类型色谱柱在同一梯度条件下比较,分别为ACQUITY UPLC BEH C18柱(1.7 μm,2.1×100 mm)、ACQUITY UPLC BEH HILIC柱(1.7 μm,3.0×100 mm)和ACQUITY UPLC HSS T3柱(1.8 μm,2.1×100 mm)。图1显示了3种色谱柱的保留时间,强极性农药在BEH C18柱上的保留时间最短,大部分强极性农药的保留时间均在1 min以内,保留时间密集;HILIC柱检测得到的色谱图保留时间大多在1~2 min,保留时间也比较密集,其中乐果、甲胺磷的保留时间均为1.47 min,涕灭威、氧乐果的保留时间均为1.49 min;强极性农药经HSS T3柱的分离,获得到有效分离,色谱图峰形都较好。对比3种色谱柱的检测结果,HSS T3柱分离检测的效果最好。强极性农药在BEH C18柱和HILIC的保留时间都集中在1~2 min,形成离子对时容易相互竞争,影响定量的结果,还容易受到基质的干扰,并且峰形对称性差。HSST3柱是硅胶基体C18柱,能够增强极性化合物的保留,减弱疏水性化合物的保留,9种化合物的分离效果见图1中HSS T3色谱图部分,保留时间具体是乐果5.76 min、甲胺磷1.54 min、多菌灵5.56 min、涕灭威6.21 min、氧乐果3.29 min、敌百虫5.38 min、噻虫嗪5.28 min、涕灭威砜4.85 min和涕灭威亚砜4.27 min;色谱峰峰形对称性比前两种好,积分定量更加准确。
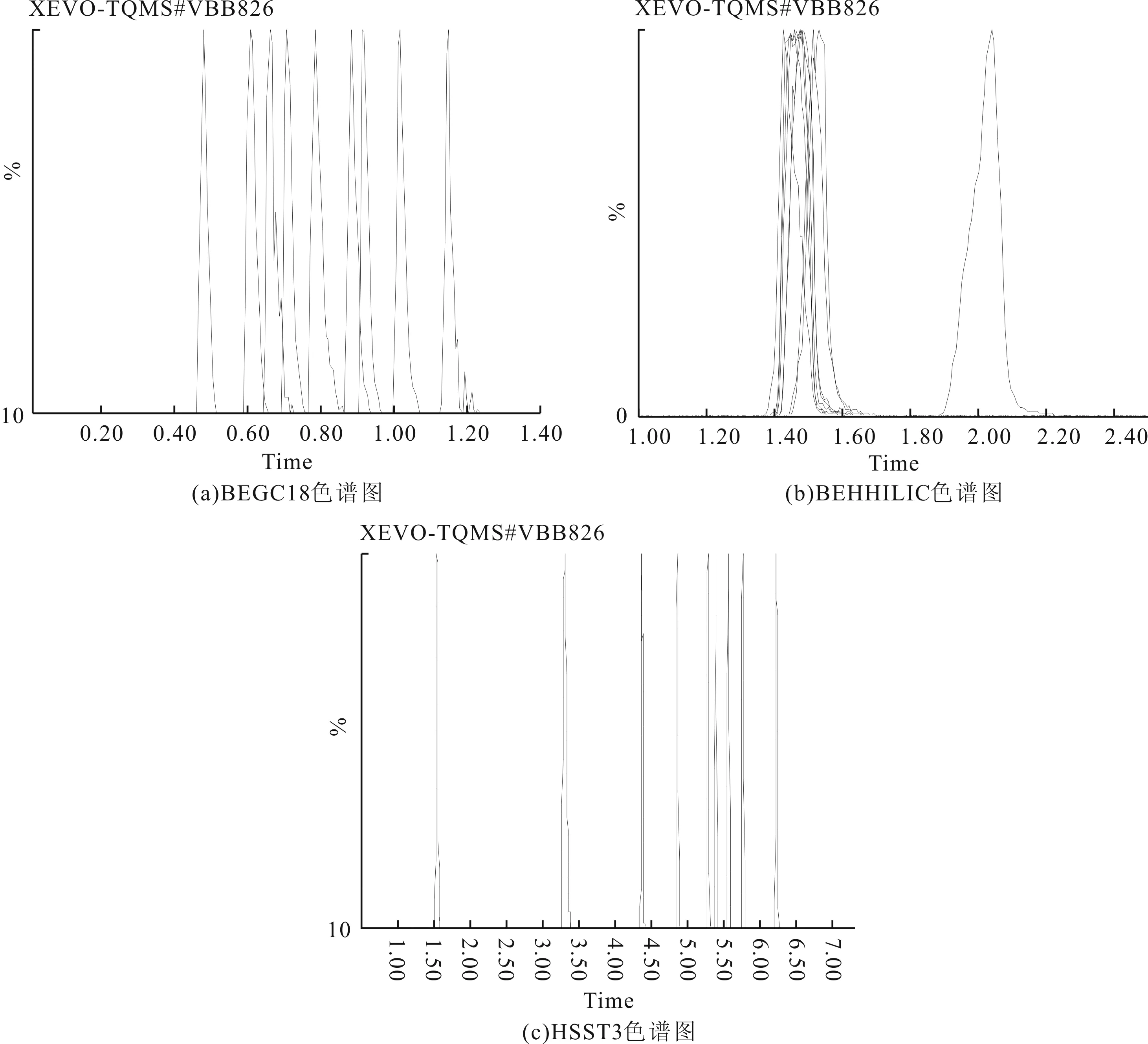
图1 9种极性农药在3种色谱柱上的色谱图
甲醇配制9种药物浓度0.2 μg/mL的混合标准溶液,质谱注射泵直接进入质谱做一级质谱MS扫描,优化毛细管电压寻找母离子;优化碰撞能量,选择响应高和干扰小的子离子作为定量离子。电离模式、定量离子对、定性离子对、碰撞能量及锥孔电压见表1。
3.2 净化方法的选择及优化
3.2.1 净化剂的优化
基质分散剂C18可以去除茶叶提取液中脂肪、甾醇和维生素等非极性物质,降低干扰和减小基质效应,提高分析结果准确性和重现性;PSA 可有效去除茶叶中脂肪酸、有机酸和糖类等干扰杂质;茶叶样品经提取色素比较重,而GCB可以消除色素对实验结果的干扰和质谱离子化的影响。按照步骤2.2节处理茶叶样品,添加水平为100 μg/kg。另设置4组不同的吸附剂的对照实验,吸附剂组合分别为400 mg PSA、400 mg C18、400 mg PSA和200 mg GCB、400 mg C18和200 mg GCB,同时加入1.2 g MgSO4,每组设置6个平行样,基质标准曲线校正,平均回收率见表2。结果显示,净化剂为PSA和C18对茶叶基质中的所有目标农药的平均回收率没有差异性,大部分目标物的回收率均低于70%;而当加入GCB时,茶叶净化液接近无色,其中甲胺磷、多菌灵、噻虫嗪、涕灭威砜和涕灭威亚砜回收率增加明显;PSA、C18和GCB同时加入时,9种化合物平均回收范围为76.48%~99.57%,符合农药残留分析要求。因此,本实验选择PSA、C18和GCB组合为茶叶基质的净化剂。
3.2.2 净化方法的选择
实验同时对比较了固相萃取法和QuEChERS法的净化效果,按照步骤2.2节处理茶叶样品,添加水平为50 μg/kg,基质标准曲线校正,平均回收率见表2。数据显示,QuEChERS法和固相萃取法处理后,9种化合物平均回收范围分别为76.48%~99.57%和63.15%~87.70%,大部分化合物差异不明显,但敌百虫、噻虫嗪和涕灭威砜已经明显低于70%。固相萃取小柱的价格比较贵,综合考虑选择QuEChERS为本实验的净化方法。

表2 不同净化条件下得到的回收率(n=6)
3.3 基质效应
在UPLC-MS/MS检测复杂样品时,通常会受到样品基质的干扰,影响目标物的离子化,产生增强或抑制效应,这种现象称为基质效应(matrix effect,ME),基质效应(ME)=基质校正曲线斜率/溶剂校正曲线斜率)[17]。溶剂相配制标准溶液系列和空白茶叶样品配制相同浓度的基质标准溶液系列,UPLC-MS/MS测定,表3结果显示:涕灭威、涕灭威砜、多菌灵、甲胺磷的ME比值在0.8~1.2范围内,为弱基质效应;乐果、氧乐果、噻虫嗪、敌百虫、涕灭威亚砜的ME比值在1.2~1.5或0.5~0.8范围内,为中等基质效应。茶叶基质中9种目标物存在不同程度的基质效应,选择空白茶叶基质匹配标准溶液进行定量分析。

表3 空白茶叶的回归方程、相关系数、基质效应、检出限和定量限
3.4 线性范围、检出限和定量限
采用基质匹配标准工作溶液绘制标准工作曲线,浓度范围在0.005~0.1 μg/mL,UPLC-MS/MS 检测,以各组分的色谱峰面积对基质标准溶液浓度进行线性回归,表3数据显示,相关系数(r)均≥0.997,线性关系良好。检出限(LOD)以定量离子的3倍以信噪比(S/N=3)计算,定量限(LOQ)以定量离子的10倍信噪比(S/N=10)计算[18]。9种强极性农药的LOD在0.09~1.57 μg/kg,LOQ在0.30~5.22 μg/kg。检出限和定量限均低于食品安全国家标准GB23200.13-2016《茶叶中448种农药及相关化学品残留量的测定液相色谱-质谱法》中的检出限及定量限[19]。
3.5 准确度和精密度
实验选择4个不同浓度水平5 μg/kg、10 μg/kg、50 μg/kg和100 μg/kg的添加回收率实验,每个浓度水平进行6次重复,基质标准曲线校正,平均回收率见表4。9种强极性农药的平均回收率在74.47%~101.9%之间,相对标准偏差(RSD%)在0.34%~12.71%之间。准确度和精密度符合《中华人民共和国农业部公告第2386号》的要求,表明9种强极性农药在茶叶油基质中的分析检测方法可行。
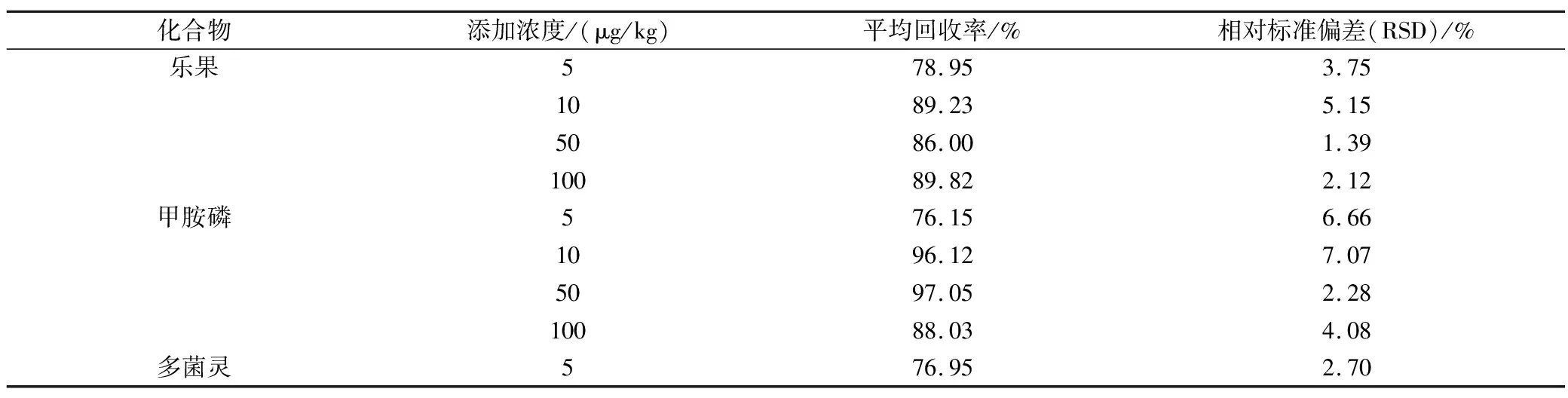
表4 空白茶叶基质中4种不同添加浓度目标物的平均回收率及相对标准偏差(n=6)
3.6 实际样品分析
采用本方法对市场和茶园上购买的5种不同批次的63个茶叶样品进行分析检测,9强极性农药均未检测出。
5 结论
本实验探究得到了适用于检测茶叶中强极性农药的色谱条件,采用QuEChERS前处理技术对茶叶进行简单、高效的前处理,用UPLC-MS/MS分析检测。本方法步骤简单易操作、试剂用量小、回收率好、精密度高,且有效除去茶叶中基质干扰,同时检出限和定量限满足于农药残留检测要求。因此,本方法适用于茶叶中涕灭威、涕灭威亚砜、涕灭威砜、氧乐果、乐果、甲胺磷、敌百虫、多菌灵、噻虫嗪9种强极性农药的检测。
