超高效液相色谱三重四极杆质谱联用测定食品汤汁中的毒品
2021-02-10罗达龙
王 华,罗达龙,覃 蓝
(梧州市食品药品检验所,广西梧州 543002)
苯丙胺(AMP)、甲基苯丙胺(MA)、3,4-亚甲基二氧基甲基苯丙胺(MDMA)、3,4-亚甲二氧基-N-乙基-苯丙胺(MDEA)和3,4-亚甲基二氧基苯丙胺(MDA)为具有苯丙胺结构的物质,该类化学组分具有中枢兴奋的作用,服用后会产生致幻、精神依赖性、食欲不振和拟交感能效应等药理与毒理学特征。在我国,该类物质是受管制的精神活性物质,故滥用潜力较大[1]。氟胺酮(2F-DCK)为氯胺酮苯环上的氯元素被氟元素所取代,在我国氟胺酮不属于管制类药物,在公安侦办的涉毒案中也屡屡查获该物质[2-4]。王世玉等[5]于1987年报道了氟胺酮的合成方法,并证明了化合物氟胺酮具有与氯胺酮类似的麻醉作用。氟胺酮能被归为麻醉活性物质的一种,也存在被滥用的潜力。
为保障人们舌尖上的安全,有必要建立一种操作简单、定性/定量准确、测试灵敏度高的检测方法。对于上述物质目前可用的方法有快检法、高效液相色谱法(High Performance Liquid Chromatography,HPLC)和动态液相微萃取(Gas Chromatography/Mass Spectrometry in the Selected Ion Monitoring Mode,GC/MS-SIM)等方法。由于火锅汤中存在的基质种类较多,如净化不完全会使杂质带入系统,产生基质干扰,影响试验回收率,甚至会引起检验结论判断错误和仪器耗损严重。
本研究将乙腈作为提取溶液,使用Captiva EMRLipid固相萃取柱进行净化,并结合UPLC-MS/MS、ACE C18-苯基色谱柱,通过多反应监测(Multiple Reaction Monitoring,MRM)对火锅汤汁中的AMP、MA、MDMA、MDEA、MDA和2F-DCK进行检测。
1 材料与方法
1.1 仪器与试剂
安捷伦超高效液相色谱仪1290-三重四极杆液质联用仪6470(美国安捷伦公司),超纯水器(赛多利斯Arium Comfort Ⅱ);超声仪;高速冷冻离心机;Captiva EMR-Lipid固相萃取柱(1 mL,40 mg,安捷伦科技有限公司)。
乙腈(质谱级,欧姆尼公司);检验用水为经赛多利斯Arium Comfort Ⅱ纯水机制备的去离子水;苯丙胺类混合标准品购自于安捷伦科技有限公司,质量浓度100 μg/mL;氟胺酮标准品质量浓度100 μg/mL,购买于阿尔塔公司。
1.2 色谱条件
分析用色谱柱:ACE C18-苯基柱(2.1 mm×100 mm,2 μm);流动相:0.2%甲酸-15 mmol/L乙酸铵(A)-乙腈(B);流速:0.35 mL/min;进样体积:2 μL;柱温箱温度:35 ;液相色谱流动相洗脱程序:0~ 1.50 min,10% ~ 20% B;1.50~ 3.50 min,20%~35% B;3.51~4.50 min,95% B,后运行时间:1.00 min。
1.3 质谱条件
ESI离子源正模式采集,干燥气体温度与干燥气体流量:150 ,12 L/min;鞘气加热温度与鞘气流量:350 ,12 L/min;喷雾器压力:25 Psi;毛细传输管电压:3 500 V;化合物离子对、碰撞能量AMP:136→ 119(5 V)91(17 V);MA:150→ 91(17 V)119(5 V);MDMA:194→ 163(9 V)105(25 V);MDEA:208→105(25 V)163(9 V);MDA:180→163(5 V) 135(18 V);2F-DCK:222→163(20 V) 109(40 V)。
1.4 标准溶液的制备
精密吸取苯丙胺类标准品和氟胺酮标准品各1 mL,置于10 mL经标定合格的容量瓶中,用乙腈溶剂稀释标品溶液至刻度,得到浓度为10 μg/mL的对照品保存液。用乙腈将对照品保存液进行梯度稀释,得到浓度分别为S1:0.02 ng/mL、S2:0.1 ng/mL、S3:0.2 ng/mL、S4:1.0 ng/mL和 S5:2.0 ng/mL的标准溶液。
1.5 供试样品的制备
取火锅汤样品,在9 000 r/min,4 条件下离心4 min,吸取离心后的上层清汤液体5.00 mL至25.00 mL容量瓶,用乙腈溶剂定容至刻度,摇匀。取稀释液约3 mL,在15 000 r/min,4 条件下离心3 min。吸取经离心后的上清液1 mL过EMR净化柱,收集后用有机滤膜过滤,上机测定。
2 结果与分析
2.1 线性关系考察
取本文“1.4”节中配制的S1~S5,按“1.2~1.3”条件,上仪器进行测定,通过仪器采集软件采集各物质的峰面积。以配制的标准曲线浓度(x,ng/mL)为横坐标,采集所得的峰面积(y)为纵坐标,使用仪器自带工作站进行线性回归,各物质线性回归方程如下。AMP:y=34 524.298 821x-755.830 859,r=0.999 2;MA:y=53 280.726 060x-986.025 191,r=0.999 6;MDMA:y=55 132.373 168x-1 236.319 062,r=0.999 7;MDEA:y=34 724.761 291x-761.259 699,r=0.999 2;MDA:y=22 939.079 908x-538.973 499,r=0.999 2;2F-DCK:y=4 1025.108 859x-765.748 774,r=0.999 6,各物质在线性浓度范围0.02~2.00 ng/mL时,物质线性的相关系数均不小于0.995 0。
2.2 回收率试验
将标准溶液加入至空白火锅汤样品中,采用本文“1.5”节方法进行处理,测定AMP、MA、MDMA、MDEA、MDA和2F-DCK 6个物质的回收率,6个物质在0.1~5.0 ng/mL的加标水平下,样品的回收率为83.2%~94.0%,RSD为3.1%~4.6%,见表1。

表1 基质加标回收率结果(n=3)
2.3 定量限与检出限试验
取本文“1.4”节下的标准溶液,进行级别稀释,按“1.2色谱条件、1.3质谱条件”进样测定。当工作站计算被测定物质的信噪比不小于10∶1时,即为定量限;当工作站计算被测定物质的信噪比不小于3∶1时,即得出检出限。由表2可知,6个物质的定量限均为0.1 ng/mL,检出限均为0.03 ng/mL。

表2 定量限与检出限试验结果
2.4 仪器精密度试验
取本文“1.4”节下的标准溶液S3,按“1.2~1.3”条件连续进样检测6次,各物质峰面积的RSD见表3。AMP:RSD=1.5%;MA:RSD=1.6%;MDMA:RSD=1.4%;MDEA:RSD=1.1%;MDA:RSD=1.6%;2F-DCK:RSD=1.6%,表明仪器精密度良好。
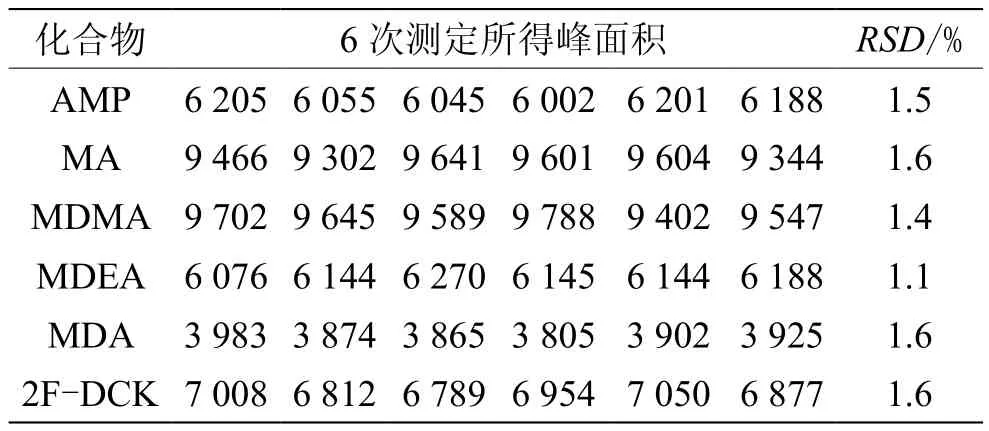
表3 精密度试验结果(n=6)
2.5 色谱柱与流动相的考察
方法考察中选择水与乙腈体系、0.2%甲酸水溶剂与乙腈混合体系、0.2%甲酸-15 mmol/L乙酸铵与乙腈溶剂混合体系等组成进行流动相适用性考察,分别在C18色谱柱与C18-苯基柱进行试验。结果发现在0.2%甲酸-15 mmol/L乙酸铵-乙腈作为流动相时,能有效促进6个化合物的电离,6个化合物的峰面积达到最大,且得到更优的灵敏度。而在C18-苯基柱上测定6个化合物得到的色谱峰峰形优于C18色谱柱。因此,选择0.2%甲酸-15 mmol/L乙酸铵-乙腈作为本次试验的流动相,选用C18-苯基柱作为本试验的色谱柱。6个化合物的色谱图见图1。

图1 化合物色谱图
2.6 样品测定
购买市售的火锅汤汁共15份按“1.2~1.5”条件进行前处理并测定,结果15份样品中均未检出AMP、MA、MDMA、MDEA、MDA 和 2F-DCK 6种物质。
3 结论
本研究建立了运用Captiva EMR-Lipid固相萃取柱对火锅汤中的基质进行净化前处理,通过使用液质联用仪的多反应监测模式,结合C18-苯基色谱柱对苯丙胺、甲基苯丙胺、3,4-亚甲基二氧基甲基苯丙胺和3,4-亚甲二氧基-N-乙基-苯丙胺、3,4-亚甲基二氧基苯丙胺和氟胺酮进行定性定量检测。从被测样品的前处理到检验完成整个试验耗时约30 min。本方法具有操作简单、快速、定性与定量准确及灵敏度高等特点,能用于日常检验。本方法使用的固相萃取柱具有高选择性,能有效去除样品基质中的脂质,所含技术为体积排阻和疏水性相互作用的原理对脂质进行净化。脂质的去除能减少对质谱电喷雾离子化的影响,使待测组分在分析过程中不受干扰,以进一步提高检测方法的稳定性与数据的可靠性,减少了仪器的维护成本。
