常染色体显性遗传Ⅳ型胶原相关肾病研究进展
2021-02-06胡宁宁杨怡青戴选彤林芙君蒋更如
胡宁宁 杨怡青 戴选彤 林芙君 蒋更如
Ⅳ型胶原是构成肾小球基底膜(glomerular basement membrane, GBM)的主要成分,为GBM中其他细胞外基质提供网状骨架支撑。Ⅳ型胶原有6种胶原链,即α1~α6,分别由COL4A1~COL4A6基因编码,α1-α1-α2链和α3-α4-α5链分别互作聚合形成Ⅳ型胶原α112和α345 两种三聚体。α1-α1-α2链在肾脏发育过程中被交联程度更高、抗蛋白水解能力更强的α3-α4-α5链所替代,因而成熟的GBM中仅存在Ⅳ型胶原α345三聚体。近年来,学者基于发病机制,将编码Ⅳ型胶原α3-α4-α5链的3个基因COL4A3、COL4A4、COL4A5任一突变导致GBM结构和功能异常的相关遗传性肾小球疾病统称为Ⅳ型胶原相关肾病(collagen Ⅳ-related nephropathy)[1],其疾病谱包括Alport综合征(Alport syndrome, AS)、薄基底膜肾病(thin basement membrane nephropathy, TBMN),以及由于COL4A3或COL4A4或COL4A5基因突变所致的局灶节段性肾小球硬化(focal segmental glomerulosclerosis, FSGS)。随着二代测序(next generation sequencing,NGS)在临床应用的普及,儿童和成人CKD患者中Ⅳ型胶原相关肾病的检出率显著提高。值得关注的是,COL4A3或COL4A4基因杂合突变导致的常染色体显性遗传Ⅳ型胶原相关肾病的实际发病率并不低,约占所有CKD患者的0.3%~1.0%[2-4]。因此,重视该类患者的早期诊断并行积极治疗对人群CKD的整体防治具有重要意义。
1 常染色体显性遗传Ⅳ型胶原相关肾病的临床疾病谱
由COL4A3或COL4A4基因杂合突变导致的常染色体显性遗传Ⅳ型胶原相关肾病临床表型的异质性较强。相关异质性和诊断分型见图1。患者大多仅表现为孤立性镜下血尿或合并非肾病综合征程度和微量蛋白尿,部分患者在血尿、蛋白尿的基础上发生肾功能减退,另有少部分患者在平均30岁前即进展为终末期肾病(end stage renal disease, ESRD)且合并眼、耳等肾外表现。肾脏病理亦呈异质性,从单纯的GBM变薄到GBM发生厚薄不均、致密层撕裂分层样改变,部分可合并FSGS样改变。
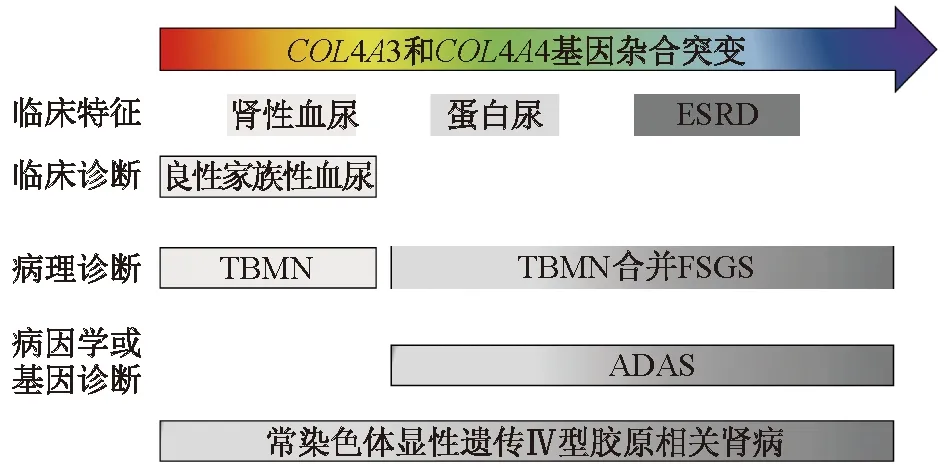
ADAS为常染色体显性遗传Alport综合征图1 常染色体显性遗传Ⅳ型胶原相关肾病临床表型异质性和诊断分型[5]
既往根据肾脏病理形态命名的TBMN是导致儿童和成人持续性镜下血尿的常见病因,在人群中所占比例高达1%[6]。TBMN以GBM弥漫变薄为病理特征。1995年WHO提出成人的GBM阈值为250 nm,2~11岁儿童为180 nm[7]。1966年,McConville等[8]首次公布了一组以持续性血尿为主要表现,而无高血压、水肿、蛋白尿、肾功能不全的患者资料;其中,行肾活组织检查(简称活检)的患者,仅个别患者系膜细胞轻度增生;而家系调查发现大部分患者有血尿家族史, 故称为“良性家族性血尿”。Voskarides等[9]研究发现,TBMN患者的肾脏预后并非均为良好。患者在疾病早期多为肾性血尿,肾活检仅见GBM弥漫变薄;30岁后约半数患者出现显性蛋白尿,在GBM弥漫变薄的基础上合并FSGS;50岁后约2/3患者发生不同程度的肾功能减退。1996年,Lemmink等[10]通过基因连锁分析首次明确COL4A4基因为TBNM的致病基因。据统计,约40%~69%的TBMN由COL4A3或COL4A4基因杂合突变引起[11-12]。近期国外一项meta分析[13]提示,在COL4A3和(或)COL4A4基因杂合突变的TBMN和(或)ADAS患者中,约29%发展为CKD,15%在约53岁时进展为ESRD。因此,TBMN这一疾病命名并不能完全反映疾病的发展进程。
此外,随着NGS的推广应用,COL4A3、COL4A4、COL4A5基因目前被证实是遗传性FSGS的首要致病基因。在家族性FSGS和散发性FSGS中,COL4A3、COL4A4、COL4A5基因突变分别约占47.3%和10.0%[14]。不同于经典的AS,COL4A3、COL4A4、COL4A5基因突变导致的FSGS患者起病较早,多数有肾性血尿、GBM异常、家族史,30岁以后逐渐发展成ESRD,无明显的眼、耳等AS特征性肾外表现。光学显微镜下可见典型的节段性硬化,电子显微镜(简称电镜)下见足突消失,但GBM无AS肾脏典型的病理表现(肾小球GBM厚薄不均,致密层撕裂、分层、虫蚀样或篮网样改变)。COL4A3、COL4A4、COL4A5基因突变导致FSGS的机制假说:①GBM病变继发足细胞损伤。一方面,COL4A3、COL4A4、COL4A5基因突变引起正常Ⅳ型胶原α345三聚体合成与胞外分泌减少,导致GBM抵抗肾小囊内静水压等机械压力的能力减弱,稳定性降低[15]。此外,GBM内层粘连蛋白(laminin)α1-α5同时发生代偿性合成增多并积聚,使GBM发生改变进而影响邻近足细胞的黏附,导致足细胞骨架结构异常[16]。另一方面,足细胞表达的Ⅳ型胶原受体α1β1整合素、α2β1整合素、盘状结构域受体(discoidin domain receptor, DDR)1和DDR2可通过识别GBM中异常Ⅳ型胶原链介导足细胞内多种促炎症因子和促纤维化因子[如TGF-β1、结缔组织生长因子、基质金属蛋白酶(MMP)2和MMP9等]表达上调进而引起足细胞损伤。②COL4A3、COL4A4、COL4A5基因突变引起的细胞内效应直接导致足细胞损伤。COL4A3、COL4A4、COL4A5基因突变可导致Ⅳ型胶原α3-α4-α5链错误折叠并在足细胞内聚集,诱导内质网应激和未折叠蛋白反应(unfolded protein response,UPR),导致足细胞凋亡等细胞损伤。2014年,Pieri等[17]在携带COL4A3-G1332E基因错义突变的患者和小鼠足细胞中均观察到内质网中滞留的α3突变体,并检测到UPR激活的相关标志物。
除了TBMN、COL4A3和(或)COL4A4基因突变导致的FSGS外,常染色体显性遗传Ⅳ型胶原相关肾病还包括ADAS。ADAS由Shaw等[18]于1976年首次报道。从现有2个样本量相对较大的ADAS临床队列[19-20]统计结果看,这类ADAS患者发生蛋白尿和ESRD的时间相较由COL4A5基因半合子突变所导致的X连锁显性遗传型AS(X-linked AS,XLAS)和由COL4A3或COL4A4基因纯合或复合杂合突变所导致的常染色体隐性遗传型AS(autosomal recessive AS,ARAS)明显延迟(蛋白尿和ESRD发生的平均年龄分别为19和70岁)。其中,约24.3%的患者在平均年龄50岁前发生ESRD,合并肾外表现的比例极低,仅4%的患者发生听力丧失和眼部异常。电镜研究数据[5]显示,44%的患者存在基底膜变薄,但无撕裂、分层,仅有少部分患者达到AS的诊断标准。故学者建议,将携带COL4A3或COL4A4基因杂合致病突变的患者诊断为常染色体显性遗传Ⅳ型胶原相关肾病[21]。但也有学者认为,应该将所有尿液检测异常的COL4A3或COL4A4基因杂合突变患者均诊断为ADAS,旨在引起医师和患者的重视,对疾病进行随访和干预,避免肾功能损伤或发生延缓其进展[5]。
2 常染色体显性遗传Ⅳ型胶原相关肾病的基因与表型关联分析
基因诊断对常染色体显性遗传Ⅳ型胶原相关肾病的确诊、家系内患病亲属的筛查,以及产前诊断至关重要。通过基因诊断及时明确病因有助于对疾病进行针对性治疗从而延缓肾功能损伤的进展,避免或减少糖皮质激素和(或)免疫抑制剂的使用。
随着NGS在临床应用的普及,越来越多的COL4A3、COL4A4、COL4A5基因突变位点被检出。目前,在人类基因突变数据库(human gene mutation database, HGMD)[22]和荷兰莱顿基因突变数据库(Leiden open variation database, LOVD)[23]中共收录了近2 000个COL4A3、COL4A4、COL4A5基因突变位点,其中COL4A5基因突变占68%,COL4A3、COL4A4基因突变占32%。上述COL4A3、COL4A4、COL4A5基因突变位点中同时存在于2个或以上独立家系或患者的比例仅为19%,其他突变位点多为个案报道。此外,通过NGS或其他基因检测手段,大量COL4A3、COL4A4、COL4A5基因的新发突变和不明意义变异位点(variant of undetermined significance, VUS)还在不断被发现[24]。除借助生物学信息学分析判断这些位点的致病性外,还可利用患者自身细胞[25](如提取尿液中足细胞,或将皮肤成纤维细胞诱导为足细胞,或构建类器官等)、体外工具细胞和动物模型开展各项体内、体外功能实验。美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics,ACMG)发布的《ACMG遗传变异分类标准与指南》[26]对新发突变的致病性列出了详细的判断标准,以帮助明确COL4A3、COL4A4、COL4A5基因新发突变和VUS位点的致病性分类。
同一基因在不同区域发生不同类型突变可导致轻重不一的临床表型,即基因型与临床表型关联(genotype-phenotype correlation)[27]。目前未发现Ⅳ型胶原相关肾病的热点突变区域,疾病的基因型与临床表型的关联证据主要来自于男性XLAS患者。3项大样本的研究[28-30]结果显示,携带COL4A5基因大片段重排或缺失或插入、单个碱基突变导致的无义突变或移码突变或剪接位点突变是较常见的男性XLAS患者与临床表型关联的基因型,且患者的临床表现较重。对于COL4A5基因氨基端非胶原区(7S)、含大量甘氨酸(Glycine,Gly)-X-Y重复结构的胶原区(X、Y代表除Gly外其他氨基酸)和羧基端非胶原区3个结构域内单个碱基突变(如错义突变、剪接位点或内含子突变、不引起移码的小片段缺失或插入)是否导致临床表型和肾脏疾病预后有所差异,目前的观点尚不一致。2000年,Jais等[28]对携带COL4A5基因突变的195个家系的共401例男性患者进行分析,结果提示:携带大片段重排、无义突变、移码突变的患者于30岁之前进展至ESRD的概率均为90%;携带剪接位点突变、错义突变的患者肾损伤相对较轻,30岁前进展至ESRD的概率分别为70%、50%。2002年,Gross等[29]对267例XLAS患者进行的meta分析结果提示:①大片段重排、无义突变、移码突变、剪接供体位点突变或羧基端非胶原区发生突变患者进展至ESRD的中位年龄为19.8岁;②21~47号外显子Gly错义突变、框内突变和涉及剪接受体位点突变患者进展至ESRD的中位年龄为25.7岁;③1~20号外显子Gly错义突变患者进展至ESRD的中位年龄为30.1岁。Bekheirnia等[30]于2010年对来自175个家庭的681例男性XLAS患者进行分析,结果提示,携带错义突变、剪接位点突变、截短突变、缺失突变的患者进展至ESRD的中位年龄分别为37、28、25、22岁。
女性XLAS患者发生的X染色体失活(里昂化,lyonization)为根据基因型判断临床表型和肾脏预后带来一定难度[31]。XLAS临床症状的严重程度和肾脏预后还与携带修饰基因位点的多态性,COL4A3、COL4A4、COL4A5的双基因或三基因突变,以及合并其他原发或继发性肾脏病和环境因素等有关[32]。
目前,关于ARAS的基因型与临床表型相关性尚无定论。据Storey等[33]报道,在ARAS中存在COL4A3、COL4A4基因截短突变的患者肾功能衰竭发生时间较早。然而,Oka等[34]未在ARAS中发现基因型与临床表型的相关性。
相较XLAS和ARAS,常染色体显性遗传Ⅳ型胶原相关肾病的不同家系间,以及同一家系内不同患病亲属间均可出现显著的临床异质性[5]。2项样本量较大的ADAS研究[19-20]均未发现显著的基因型与临床表型的关联。Andreas等[35]对74例进展至ESRD的COL4A3、COL4A4基因杂合突变患者进行meta分析后发现,54例错义突变的患者进展至ESRD的平均年龄为55.2岁,20例非错义突变(导致翻译提前终止的缺失、重复、剪接位点突变)患者进展至ESRD的平均年龄为47.1岁。两类患者进展至ESRD的年龄差异具有统计学意义。
针对上述局限,最近Kamura等[36]基于纳米荧光素酶片段互补技术(split-nanoluciferase binary technology)建立了一种体外实时定量分析Ⅳ型胶原α345三聚体的聚合稳定性的分子诊断新方法。由Ⅳ型胶原α3-α4-α5单链在足细胞内互作聚合形成并分泌至胞外的Ⅳ型胶原α345三聚体是构成GBM中Ⅳ型胶原网的核心骨架分子。该研究挑选了9个基因型与临床表型关联确凿且导致临床表型轻重不一的COL4A5错义突变基因以验证该方法的准确性[36],发现各突变引起的Ⅳ型胶原α345三聚体聚合稳定性改变与临床表型显著相关。该团队还发现,COL4A5基因突变,如G230C,主要引起足细胞内Ⅳ型胶原α345三聚体聚合障碍,携带该突变的患者进展至ESRD的平均年龄仅21.5岁,临床表型较重。主要引起Ⅳ型胶原α345三聚体分泌水平轻度异常而不引起足细胞内Ⅳ型胶原α345三聚体聚合异常的COL4A5基因突变(如G509R、G805R和G1143S)所致的临床表型较轻,蛋白尿和ESRD发生的时间明显延迟。致病性目前尚不明确的多态性位点,如G953V,对Ⅳ型胶原α345三聚体聚合稳定性的影响与野生型相比无显著差异。
3 常染色体显性遗传Ⅳ型胶原相关肾病的治疗
目前,针对Ⅳ型胶原相关肾病的治疗进展主要集中于AS,包括药物治疗、肾脏替代治疗、肾移植术、基因和细胞治疗。常染色体显性遗传Ⅳ型胶原相关肾病患者如出现尿蛋白增多或肾损伤,以RAAS抑制剂治疗为主。对于出现大量蛋白尿或肾病综合征的患者,则予以糖皮质激素和(或)免疫抑制剂治疗。进展至ESRD患者,可行肾脏替代治疗或肾移植术。
3.1 RAAS抑制剂 2013年,国际AS专家组发表的诊治建议[37]将治疗药物分为一线和二线用药,其中一线治疗应用ACEI,这类药物包括雷米普利、依那普利等;二线治疗应用ARB和醛固酮受体拮抗剂;常用的ARB类药物包括氯沙坦、厄贝沙坦、缬沙坦等。一项大型回顾性研究[38]结果表明,早期应用ACEI可使AS患者进展至透析的时间延迟13年。2020年,一项名为EARLY PRO-TECT Alport(Early Prospective Therapy European Community Trial Alport)的Ⅲ期临床试验[39]结果表明,在AS患儿出现显性尿蛋白之前使用雷米普利能有效延缓其蛋白尿的进展和肾小球滤过率的下降,使疾病进展的风险降低近一半(风险比为0.52,95%CI: 0.19~1.39),并且尚未发现安全问题。2017年,首个关于RAAS抑制剂应用于携带COL4A3、COL4A4、COL4A5基因杂合突变患者的前瞻性研究[40]结果表明,RAAS抑制剂亦可有效延缓此类患者的肾功能损伤进展。
3.2 糖皮质激素和(或)免疫抑制剂 对于少部分表现为大量蛋白尿或肾病综合征的TBMN患者,可试用糖皮质激素。据Nogueira等[41]报道,3例表现为重度蛋白尿或肾病综合征的TBMN患者中有2例接受糖皮质激素治疗后蛋白尿缓解或显著改善。环孢素A(cyclosporine A,CsA) 为钙调神经蛋白磷酸酶抑制剂,是一种强效免疫抑制剂,可直接改变GBM的通透性,改善肾脏血流动力学,发挥降低尿蛋白的作用。1999年,Callís等[42]报道称,CsA治疗AS可显著降低尿蛋白水平,并在长期随访中显示出肾脏保护作用。2007年,Charbit等[43]使用CsA治疗9 例 AS 患者后发现,患者尿蛋白减少的同时伴有肾小球滤过率降低,并且3例患者在第二疗程结束时复查肾活检均显示有明显肾毒性。因此,目前CsA的应用仍存在争议。COL4A3、COL4A4、COL4A5基因突变导致的FSGS对免疫抑制剂反应差,治疗前进行基因诊断明确致病基因可避免不必要的免疫抑制治疗和由其导致的药物不良反应。
3.3 带来肾脏获益的新型治疗药物 新型治疗药物如甲基巴多索酮(bardoxolone methyl)和抗微RNA-21已进入临床试验阶段。甲基巴多索酮可抑制NF-κB,并诱导核因子E2相关因子(Nrf2)表达,从而发挥抑制炎症反应,诱导线粒体功能恢复,减少氧化应激的作用。此外,甲基巴多索酮已被证实可提高2型糖尿病合并CKD患者的肾小球滤过率[44],但其安全性尚存争议。尽管甲基巴多索酮可提高肾小球滤过率,但同时可能会伴随高滤过状态和肾小球高压,后者会损伤GBM,并提高尿白蛋白/肌酐比值[45]。此外,一项关于2型糖尿病合并CKD4期患者的临床研究[46]发现,与安慰剂组(1 097例)中的55例相比,甲基巴多索酮组(1 088例)共有96例患者因心力衰竭住院或死亡(风险比为1.83,95%CI: 1.32~2.55,P<0.001)。目前,一项关于甲基巴多索酮治疗AS的有效性和安全性的Ⅱ期和Ⅲ期临床试验已启动(临床试验编号:NCT03019185)。但仍需更多临床研究以指导甲基多巴索酮的应用。微RNA-21参与包括肾脏在内的多个器官的纤维化与损伤后组织修复的调节。2015年,Gomez等[47]在AS小鼠模型中发现,抗微RNA-21药物可通过刺激代谢途径阻止AS的进展,改善AS小鼠的肾脏病理损害和肾功能,使患有持续进展性肾病的小鼠中位生存时间延长>40%。目前,一项关于SAR339375(抗微RNA-21药物)治疗AS的Ⅱ期临床试验正在进行中(临床试验编号:NCT02855268)。
4 常染色体显性遗传Ⅳ型胶原相关肾病的遗传咨询
临床医师应注重对常染色体显性遗传Ⅳ型胶原相关肾病患者进行遗传咨询(遗传概率为50%),避免近亲婚配,尽可能优生、优育。对于家系中有生育意愿者可进行产前诊断,如进行胎盘植入前遗传学诊断(preimplantation genetic diagnosis,PGD),以选择遗传学健康的胚胎用于移植,阻断子代遗传,提高人口素质。
5 总结与展望
由COL4A3或COL4A4基因杂合突变所导致的常染色体显性遗传Ⅳ型胶原相关肾病是根据疾病发病机制而命名的新概念,其疾病谱包括COL4A3或COL4A4基因杂合突变引起的TBMN、FSGS和ADAS。常染色体显性遗传Ⅳ型胶原相关肾病在CKD人群中的比例约占0.3%~1%,重视该病的早期诊断对CKD防治具有重要意义。常染色体显性遗传Ⅳ型胶原相关肾病的临床表型异质性强,多数患者可进展至ESRD,可能与患者所携带的基因型、合并其他肾脏疾病或影响肾功能进展的危险因素等有关。近年来,在药物治疗AS的临床研究领域有较多进展,其研究结果可否运用于常染色体显性遗传Ⅳ型胶原相关肾病有待后续研究明确。
