玫瑰高原鳅肠道微生物多样性研究
2021-02-04彭作刚
刘 妮 彭作刚
(西南大学生命科学学院,淡水鱼类资源与生殖发育教育部重点实验室,重庆 400715)
玫瑰高原鳅(Triplophysa rosa)[1],隶属于鲤形目(Cypriniformes),条鳅科(Nemacheilidae),高原鳅属(Triplophysa),是一种小型鱼类。目前仅分布于重庆市武隆县梦冲塘,属乌江水系,是我国特有鱼类。玫瑰高原鳅常年栖息于地下暗河或溶洞中,体表色素消失,眼睛高度退化,触须、鳍条等感觉器官高度发达。目前对玫瑰高原鳅的研究集中于新种描述[1]、中枢神经系统组织学[2]、视觉器官形态结构[3]、体色白化遗传基础[4]、能量代谢[5]、系统演化[6]、遗传多样性[7]及保护遗传学[8]等方面,关于玫瑰高原鳅食性的研究未见报道。
鱼类生长所需要的能量来源于对食物的消化吸收[9]。除了有效地捕获食物外,提高消化效率、优化营养物质的摄取和吸收,对鱼类的营养需求同样重要[10]。肠道是鱼类重要的消化器官,其内存在大量微生物,所以也是一种特殊的生物生态位[11]。营养代谢、同化是宿主与肠道微生物群在肠道中的协同过程[12],肠道微生物可以产生维生素、氨基酸、消化酶、多种生长因子及其他代谢产物,从而促进鱼类的生长以及其他生理过程[13]。食性是影响肠道微生物的重要因素,不同食性的鱼类,肠道微生物存在很大差异[14,15]。因此,肠道微生物的结构组成与宿主的食性息息相关[16—19]。
Illumina Miseq测序技术具有通量高、数据完整性好、经济高效且快速的优点[20],已广泛应用于研究微生物群落结构。16S rRNA(16S ribosomal RNA)是编码原核生物核糖体小亚基的基因,突变率小且分子大小适中,是细菌系统分类学研究中最常用和最有用的分子标记。
本研究通过使用Illumina Miseq平台对玫瑰高原鳅肠道微生物16S rRNA的V3—V4区进行测序以对其进行多样性研究,分析玫瑰高原鳅肠道菌群的结构及多样性,为探索玫瑰高原鳅肠道菌群的作用、了解玫瑰高原鳅的消化特性等提供参考依据。
1 材料与方法
1.1 样品采集
2018年6月在重庆市武隆县梦冲塘采集玫瑰高原鳅,当天带回实验室。于无菌条件下解剖样本,完整取出肠道,置于2 mL离心管中,液氮速冻后保存在-80℃冰箱中待用。解剖工具均经过灭菌处理。
1.2 实验方法
使用E.Z.N.A.®soil试剂盒(Omega Bio-tek,Norcross,GA,U.S.),按照说明书对样品进行总DNA提取,使用NanoDrop2000对DNA浓度和纯度进行检测。针对16S rRNA基因的V3—V4区进行扩增,所用引物为338F(5′-ACTCCTACGGGAG GCAGCAG-3′)和806R(5′-GGACTACHVGGGT WTCTAAT-3′)[21],扩增区域长度约450 bp。扩增体系为20 μL:4 μL 5×FastPfu缓冲液,2 μL 2.5 mmol/L dNTPs,0.8 μL引物(5 μmol/L),0.4 μL FastPfu聚合酶,10 ng DNA模板。PCR反应条件:95℃预变性3min;95℃变性30s,55℃退火30s,72℃延伸45s,29个循环;最后72℃终延伸10min。2%琼脂糖凝胶电泳检测PCR产物。测序采用Illumina MiSeq测序平台进行双端测序。
1.3 数据分析
使用Trimmomatic软件对得到的原始序列进行质控,使用FLASH软件进行拼接。使用UPARSE软件(version 7.1,http://drive5.com/uparse/),根据97%的相似度对序列进行OTU聚类,并在聚类的过程中去除单序列和嵌合体。利用RDP classifier(version 2.2,http://rdp.cme.msu.edu/)对每条序列进行物种分类注释,比对Silva数据库(Release132,http://www.arb-silva.de),统计每个样本的肠道微生物在界、门、纲等分类水平的组成。使用mothur软件(version 1.30.1,http://www.mothur.org/wiki/Schloss_SOP#Alpha_diversity)计算香农指数(Shannon index)、辛普森指数(Simpson index)、ACE指数(ACE index)、Chao1指数(Chao1 index)及覆盖率(Coverage),评估肠道菌群的Alpha多样性。
使用PICRUSt菌群代谢功能预测工具对OTU丰度表进行标准化,然后通过每个OTU对应的Green Gene ID对比到COG库(Clusters of Orthologous Groups,直系同源基因簇)和KEGG库(Kyoto Encyclopedia of Genes and Genomes,京都基因与基因组百科全书),获得OTU对应的COG家族信息和KO信息,计算各COG的丰度和KO丰度。根据COG数据库的信息,可以从eggNOG数据库中解析到各个COG的描述信息,及其功能信息,从而得到功能丰度谱;根据KEGG数据库的信息,可以获得KO、Pathway、EC信息,并根据OTU丰度计算各功能类别的丰度。
2 结果
2.1 测序基本数据分析
采用Illumina Miseq测序平台对5尾样品的16S rRNA基因V3—V4区进行测序,共得到有效序列218278条,按97%相似度聚类后得到451个OTUs,共鉴定出19门、31纲、87目、146科、253属、320种。
Alpha多样性是指一个特定区域或生态系统内的多样性,是反映丰富度和均匀度的综合指标。该分析中,香农指数、辛普森指数反映菌群的多样性,香农指数值越高表明菌群的多样性越高,辛普森指数值越低表明菌群的多样性越高;ACE指数、Chao1指数反映菌群的丰富度,ACE指数、Chao1指数值越高表明菌群物种的丰富度越高;Coverage值为测序深度指数,其值越接近于1,说明测序深度越合理。Alpha多样性分析结果表明:5尾样品的丰富度与均匀度差异较大。样品TR2的香农指数、ACE指数、Chao1指数最高。5个样品的coverage值从0.998到1,表明测序深度已基本覆盖到样品中所有的物种(表1)。

表1 Alpha多样性统计Tab.1 The statistics of Alpha-diversity
随着测序深度的不断增加,稀释曲线趋于平缓,表明本实验获得的数据可以反映玫瑰高原鳅肠道微生物的绝大部分信息(图1)。5尾样品共有的OTUs有10个,样品TR3独有的OTUs最少,仅有3个,样品TR2独有的OTUs最多,为151个(图2)。
2.2 玫瑰高原鳅肠道菌群整体结构分析
基于门分类水平上的玫瑰高原鳅肠道微生物多样性分析高通量测序分析表明玫瑰高原鳅的肠道微生物主要由变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、蓝藻细菌(Cyanobacteria)、绿弯菌门(Chloroflexi)、Patescibacteria、Epsilonbacteraeota、异常球菌-栖热菌门(Deinococcus-Thermus)、螺旋体门(Spirochaetes)等组成。在样品TR1、TR2中,主要的菌门为变形菌门(93.61%和86.15%)以及少量的放线菌门(4.47%和2.07%)和拟杆菌门(1.01%和1%),此外,TR2中,还有3.28%的厚壁菌门;在TR3、TR4及TR5中,主要的菌门为变形菌门(98.67%、98.31%、97.28%)和少量的拟杆菌门(1.25%、1.15%、1.71%,图3)。
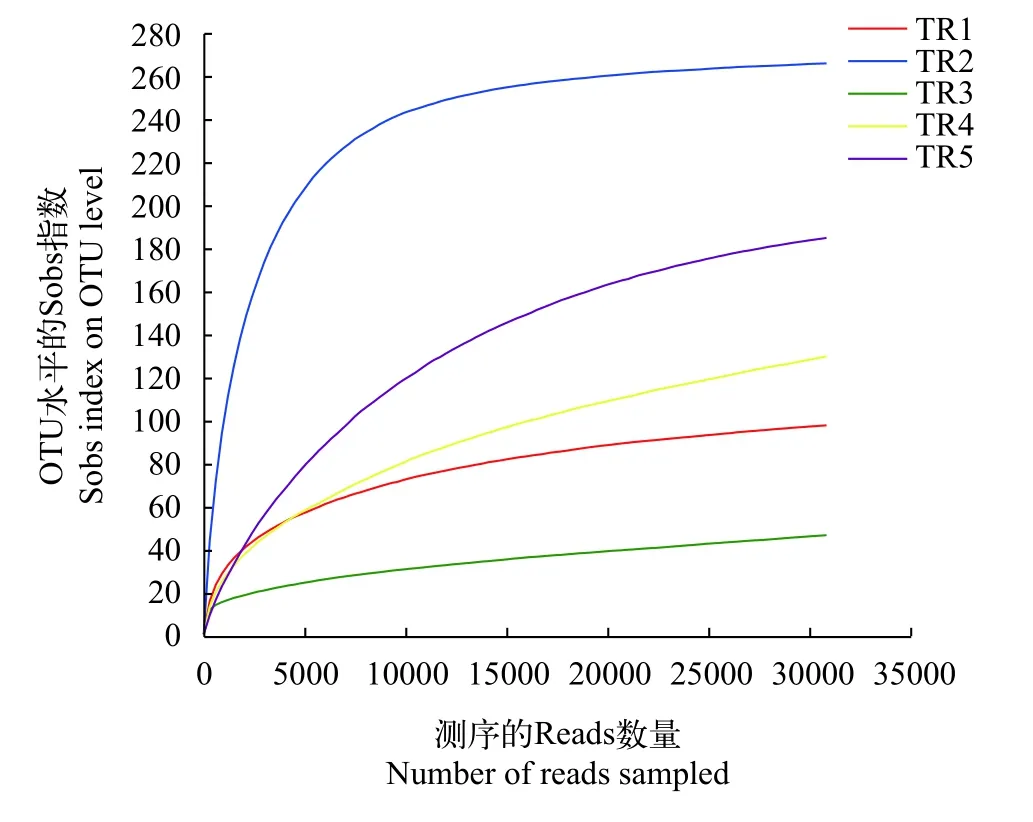
图1 样品的稀释曲线图Fig.1 The rarefaction curve of samples
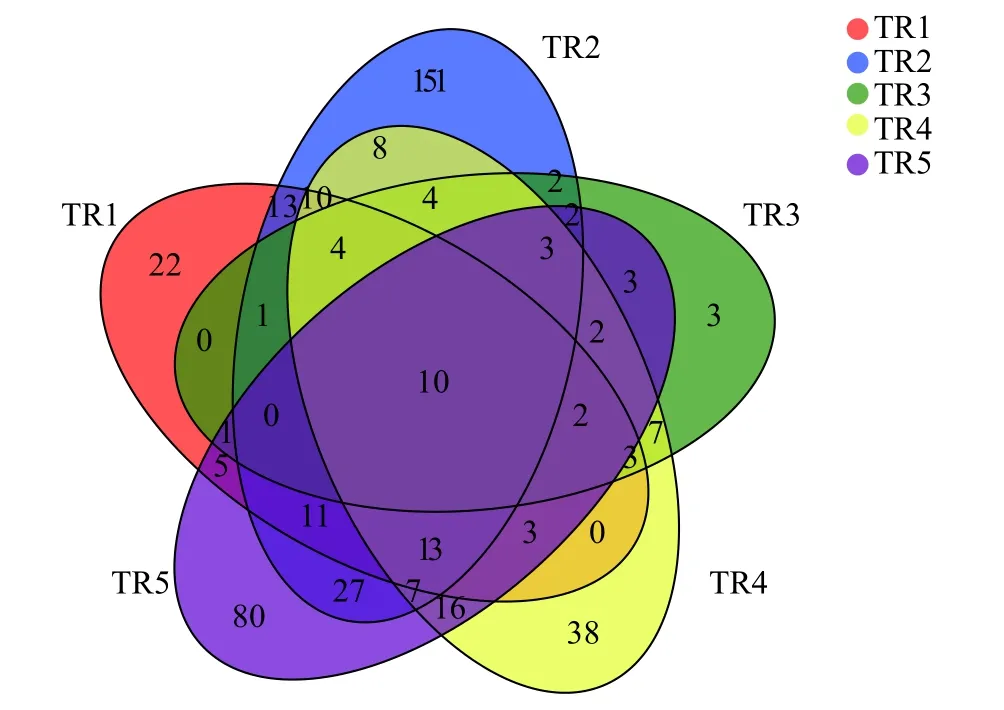
图2 各样本OTUs的韦恩图Fig.2 The venn diagram of each sample’s OTUs
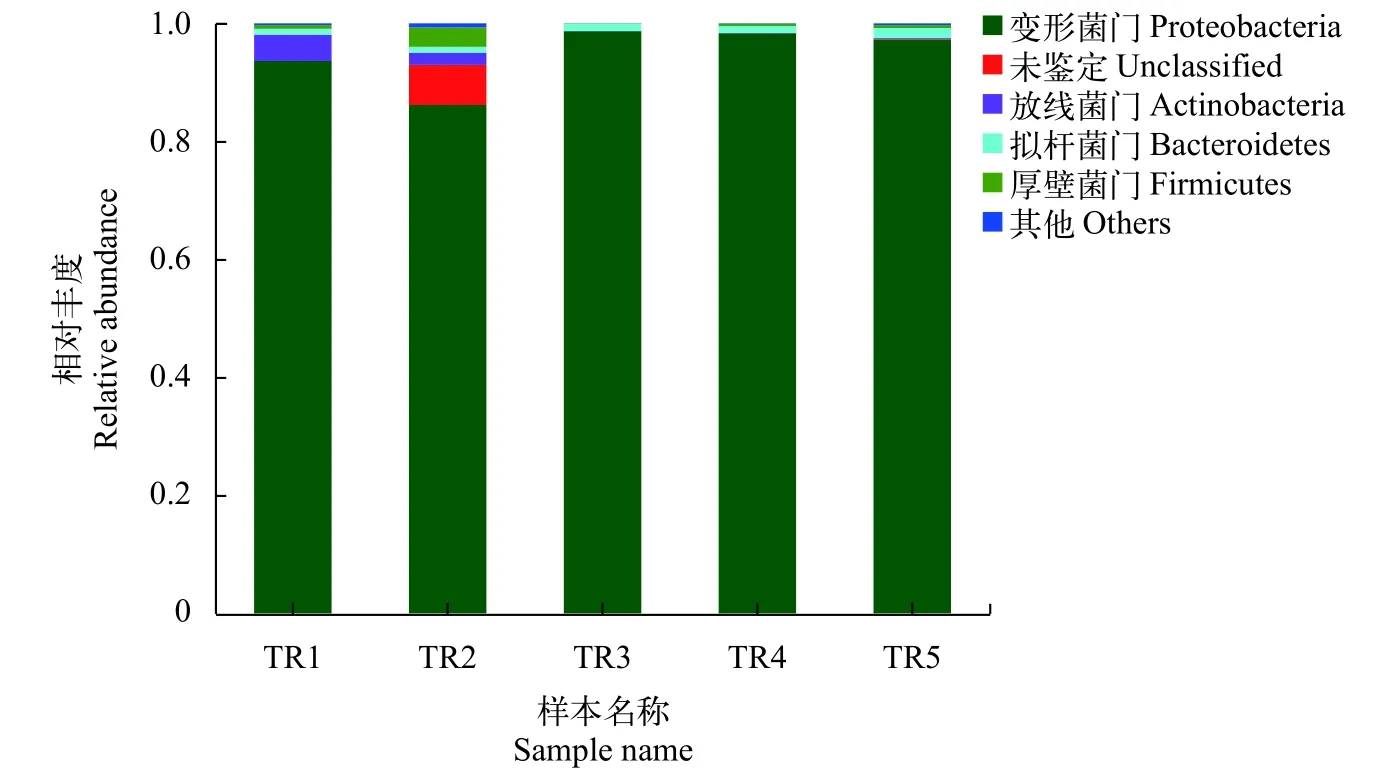
图3 基于门水平的玫瑰高原鳅肠道菌群结构Fig.3 Bacterial structure evaluated at the phylum taxonomical level in T. rosa
基于属分类水平上的玫瑰高原鳅肠道微生物多样性分析在样品TR1中,主要的菌属为气单胞菌属(Aeromonas,84.93%)、希瓦氏菌属(Shewanella,5.06%)及Paenarthrobacter(3.85%);在样品TR2中,主要的菌属为邻单胞菌属(Plesiomonas,79.09%)、罗尔斯通菌属(Ralstonia,1.76%)及芽孢杆菌属(Bacillus,1.76%);在样品TR3中,主要的菌属为气单胞菌属(65.72%)、希瓦氏菌属(22.34%)、弧菌属(Vibrio,3.35%)、假单胞菌属(Pseudomonas,3.35%)及黄杆菌属(Flavobacterium,1.24%);在样品TR4中,主要的菌属为气单胞菌属(81.11%)、希瓦氏菌属(4.24%)、弧菌属(8.98%)、黄杆菌属(1.07%)及不动杆菌属(Acinetobacter,1.67%);在样品TR5中,主要的菌属为爱德华菌属(Edwardsiella,95.87%)及黄杆菌属(1.4%,图4)。
2.3 功能预测
基于各样品KEGG比对获得的功能信息组成,共获得282个代谢功能途径。KEGG代谢通路表明:肠道微生物编码的大多数基因与新陈代谢相关,其次是与环境信息处理相关(表2)。COG功能分类统计结果表明:菌群在“氨基酸转运与代谢”和“碳水化合物运输和代谢”功能类群的相对丰度较高(图5)。
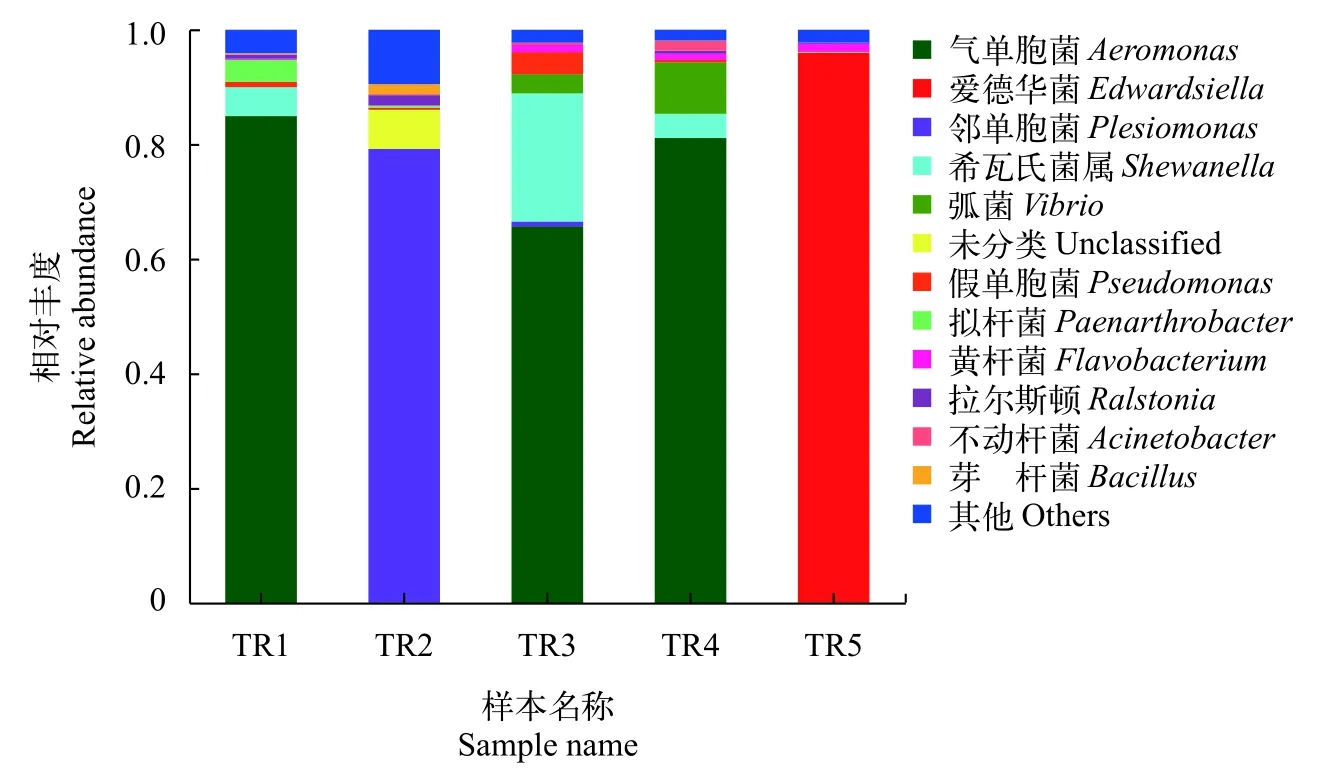
图4 基于属水平的玫瑰高原鳅肠道菌群结构Fig.4 Bacterial structure evaluated at the genus taxonomical level in T. rosa

表2 KEGG代谢通路统计Tab.2 Metabolic pathway statistics based on KEGG
3 讨论
本研究对玫瑰高原鳅的肠道微生物多样性进行了分析。Alpha多样性分析结果中,样品TR2的香农指数、ACE指数、Chao1指数最高,说明该样品中菌群最丰富。另外,5个样品的coverage指数平均值为0.999,说明样品中物种没有被检测出的概率较低,表明测序数据合理,能够反映肠道微生物菌群的结构及组成的多样性。不过本研究中5尾玫瑰高原鳅的肠道菌群组成及数量存在差异,而肠道菌群的多样性及丰度受诸多因素的影响,如年龄、性别[22,23]、进食状态[24,25]、健康状态[26]、应激反应[27]等,因此推测玫瑰高原鳅肠道菌群的差异与个体的生理状况有关。
本研究中变形菌门是玫瑰高原鳅肠道中丰度最高的菌群,其次是放线菌门、拟杆菌门和厚壁菌门,这与非洲齿鲤(Nothobranchius furzeri)[22]、虹鳟(Oncorhynchus mykiss)[28]、大西洋鲑(Salmo salar)[29]、金目鲈(Lates calcarifer)[30]等的肠道菌群相似,只是比例不同。典型洞穴鱼类墨西哥丽脂鲤(Astyanax mexicanus)肠道菌群也以变形菌门为主[31]。变形菌门、放线菌门和拟杆菌门普遍存在于鱼类肠道[32],同时也是其他动物如加拿大马鹿(Cervus canadensis)和白尾鹿(Odocoileus virginianus)[33]、雉科鸟类[34]、尖吻蝮(Deinagkistrodon)和Ptyas mucosa[35]、瑞肇大小蠹(Dendroctonus rhizophagus)[36]等物种肠道中的优势菌,表明物种之间存在共享的核心微生物群[33]。变形菌门的含量远远大于其他菌门,为玫瑰高原鳅肠道中的绝对优势菌。变形菌门是细菌中最大的一门,功能高度复杂,广泛参与宿主的营养代谢[37]。拟杆菌门可以帮助宿主降解碳水化合物、蛋白质和其他物质,促进营养物质的吸收,维持肠道稳态,促进宿主免疫系统的发育[38,39]。厚壁菌门的物种丰富,与碳水化合物代谢和吸收相关,可以产生多种消化酶帮助宿主分解各种物质,促进营养物质的吸收[40]。
样品TR1、TR3和TR4的优势菌属为气单胞菌属,属于变形菌门,该属可以分泌与致病性及环境适应有关的酶,如溶血性肠毒素、脂肪酶、蛋白酶、淀粉酶[41],可以提高食物的消化率,在消化过程中具有重要作用。玫瑰高原鳅常年生活在洞穴中,食物匮乏,具有良好的食物消化以及营养吸收能力对其生长发育极其重要。TR2中的优势菌属为邻单胞菌属,菌种为类志贺邻单胞菌(Plesiomonas shigelloides),TR5的优势菌属为爱德华菌属,菌种为鲇鱼爱德华氏菌(Edwardsiella ictaluri),类志贺邻单胞菌和鲇鱼爱德华氏菌都是常见的重要水生致病菌,在鱼体内会引起严重的肠道疾病[16,42],这两种菌在样本中的高丰度可能与该样本的健康状况有关。这可能是采样过程中,环境的突然改变破坏了鱼类肠道菌群的平衡状态,导致产生细菌性疾病[12]。
图5 玫瑰高原鳅肠道微生物的COG功能分类统计Fig.5 The COG function classification of intestinal microorganisms in T. rosa
鱼类肠道菌群组成与食性相关,如草食性鱼类肠道中主要以梭菌属(Clostridium)、柠檬酸杆菌属(Citrobacter)和纤毛菌属(Leptotrichia)为主;肉食性鱼类肠道中主要菌群是鲸杆菌属(Cetobacterium)和盐单胞菌属(Halomonas);杂食性和滤食性鱼类肠道中主要菌群为梭菌属、鲸杆菌属和盐单胞菌属[19]。本研究在玫瑰高原鳅的肠道中检测到了少量的柠檬酸杆菌属、盐单胞菌属和鲸杆菌属,推测其食性为杂食性偏肉食性。
鱼类的肠道微生物被认为对宿主的生命活动有重要影响,在宿主的营养、免疫、防御等方面发挥重要作用[43—45]。本研究中菌群功能预测的结果发现玫瑰高原鳅肠道中与碳水化合物代谢相关的菌群丰度最高,其次是与氨基酸代谢相关的菌群,表明玫瑰高原鳅的能量来源主要是纤维及蛋白质。此外还有丰度较高的与环境信息处理相关的菌群,这可能与其特殊的洞穴生活环境有关。
