基于转录组测序初步揭示天麻生长代谢的分子机制
2021-02-03刘云霞狄永国仇全雷肖舒卉谭彧文陈丽梅徐慧妮李昆志
刘云霞,狄永国,仇全雷,肖舒卉,谭彧文,陈丽梅,徐慧妮,李昆志*
1.昆明理工大学生命科学与技术学院,云南 昆明 650500
2.昭通市昭阳区农业局 种植业科,云南 昭通 657000
3.西藏波密高原藏天麻产业开发有限公司,西藏 林芝 860300
天麻是名贵传统中药材,具有息风止痉、平抑肝阳、祛风通络之功效,主治小儿惊风、癫痫抽搐、破伤风、头痛眩晕、风湿痹痛等症[1-3]。天麻为药食两用的兰科植物,具有极高的药用和食用价值,拥有巨大的潜在市场,天麻需求量大。野生天麻活性成分含量高、药效好,但产量低,不能满足实际需求,且由于人工过度采集濒临灭绝,天麻供给主要靠人工种植。因此,天麻栽培成为天麻领域的研究热点。
天麻生长经历种子、原球茎、米麻、白麻和箭麻5个生长发育阶段[4]。Kusano[5]首次报道了天麻与木材腐烂病原菌蜜环菌存在真菌菌根关系。随后的研究表明,天麻种子萌发需要萌发菌如小菇属Mycena dendrobiiL.FanetS.X.Guo 侵入种胚细胞,为天麻种子萌发提供营养物质,促进天麻种子萌发长出幼小的原球茎[6]。随着原球茎进一步生长,其前端分生组织处可直接分化出营养繁殖茎米麻。天麻可消化萌发菌和蜜环菌入侵的菌丝作为其营养来源[7-9],米麻从侵入的萌发菌和蜜环菌获得营养发育成营养繁殖茎白麻[10]。在接下来的生长中白麻的直接营养物质是蜜环菌,蜜环菌侵入白麻后,白麻消化侵入的蜜环菌菌丝获得营养后进一步长出商品麻箭麻[11]。最近,研究者通过天麻转录组学分析,阐明了天麻种子萌发的机制[12-14]。谭彧文等[15]通过转录组学的方法发现侵入到天麻体内的蜜环菌其胞外酶和抗氧化酶的差异表达基因(differentially expressed genes,DEGs)98.31%表达下调,说明侵入天麻体内的蜜环菌活性减弱,不能进一步入侵天麻,且不受到天麻胁迫作用,使蜜环菌和天麻处于共生状态,初步揭示了天麻与蜜环菌的共生机制。袁媛等[16]测出天麻的全基因组序列发现,大部分与光合作用和抗病菌相关的基因如核酸结合部位(nucleotide-binding site,NBS)类基因发生了大量丢失,同时发现天麻的线粒体基因组扩大且单子叶甘露糖结合凝集素抗真菌蛋白(gastrodia antifungal protein,GAFP)基因的数量增加,且发现天麻与蜜环菌共生关系建立的重要信号是独角金内酯,这些结果展现了完全异养植物天麻是如何通过实现广泛的基因收缩甚至丢失、扩张以及基因的新功能化来完成其独特的生长特征,更深一步地阐述了天麻与蜜环菌的共生关系。文欢等[17]也通过转录组学的方法发现了天麻内存在较完整的天麻苯丙烷类产物合成代谢通路。到目前为此,天麻各生长发育阶段的生理形态特征比较清楚,蜜环菌能入侵营养繁殖茎白麻与其共生,但共生后进一步生长为箭麻的生长代谢特征仍不清楚。
本实验通过转录组分析,在转录组水平的基础上研究箭麻和共生天麻生长代谢过程中相关差异基因表达水平变化,揭示两者间的基因表达水平的差异,以期阐释箭麻和共生天麻间的生长代谢特征,为进一步研究天麻不同生长发育阶段的生理生化代谢特征奠定基础,同时为天麻栽培提供理论指导。
1 材料与仪器
1.1 材料
选取昭通产乌天麻作为研究材料,经笔者鉴定为乌天麻Gastrodia.e lataBl.(F.gl auca) S.Chow。将蜜环菌Armillaria.mellea(Vahl) P.Kumm.接种于苹果树枝一段时间待长出蜜环菌菌丝后接种于营养繁殖茎白麻,成为共生天麻,接种3 个月长出箭麻后分别取2 种样品,立即放入液氮中冷冻后储存入-80 ℃冰箱中用于总RNA 提取。
1.2 仪器
Bio-rad CFX96 型荧光定量PCR 仪(美国伯乐公司);DHP-9502 型电热恒温培养箱(上海一恒科学仪器有限公司);heraeus mutifuge X1R 型实验室用离心机(赛默飞世尔科技有限公司)。
2 方法
2.1 RNA 提取及质量检测
采用Trizol Reagent(Invitrogen)提取箭麻和共生天麻的RNA。将约0.1 g 样品放入用液氮预冷的研钵中,磨碎后加入1 mL 的Trizol 提取液于研钵中继续研磨至研磨液呈红色透明状,于室温静置5 min 后移入1.5 mL 离心管并加入0.2 mL 氯仿振荡混匀;随后在12 000 r/min、4 ℃离心15 min 后吸取上清液至新的离心管中;在加入0.25 mL 异丙醇和0.25 mL 氯化钠与柠檬酸钠混合高盐液,混匀-20 ℃放置30 min后在12 000 r/min、4 ℃条件下离心30 min,弃上清,沉淀用1 mL(-20 ℃ 预冷)的75%乙醇清洗,随后在7500 r/min、4 ℃条件下离心5 min,弃乙醇,重复清洗2 次;然后将沉淀在室温条件下自然晾干,最后用20 μL 焦碳酸二乙酯(diethyl pyrocarbonate,DEPC)处理水溶解RNA。用分光光度计测量样品RNA 的纯度和浓度,检测RNA 样品的完整性。
2.2 转录组测序样品制备及文库建立
提取样品总RNA 后,将破碎缓冲液加入到带有Oligo(dT)磁珠富集的真核生物mRNA 中将其打断成短片段。以mRNA 为模板,用六碱基随机引物合成第1 条cDNA 链,随后加入缓冲液、RNase H、DNA 聚合酶I 和dNTPs 以合成第2 条cDNA 链。在经过QiaQuick PCR 试剂盒纯化并加EB 缓冲液洗脱之后做末端修复、加poly(A)并连接测序接头。最后用琼脂糖凝胶电泳进行片段大小选择,并进行PCR 扩增,建好的测序文库用Illumina HiSeqTM2000 进行高通量测序分析。
2.3 测序数据处理及分析
经过统计箭麻和共生天麻分别获得平均28.97 Mb 和67.73 Mb 条原始测序序列(raw reads)。对raw reads过滤低质量数据和测序引物、接头等人工序列,得到clean reads。对过滤处理得到的clean reads 进行Trinity 组装生成转录本(transcript)和单基因簇(unigene)。再将Unigene 与Swiss-Prot(瑞士-波特蛋白质序列数据库)、非冗余蛋白库(non-redundant protein sequence database,NR)、京都基因和基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)和蛋白相邻类的聚簇(cluster of orthologous groups of proteins,KOG)数据库比对,获得Unigene 功能注释信息。
2.4 表达差异分析
使用RPKM 值表示Unigene 的表达丰度。满足false discovery rate(FDR)<0.05 且log2|FC|≥2 条件的基因为差异表达基因(differentially expressed genes,DEGs)。随后对差异表达基因做基因本体论(Gene Ontology,GO)功能富集分析、KOG 注释和KEGG 注释与分类。
2.5 实时荧光定量PCR (qRT-PCR) 分析
为了验证转录组数据中基因差异表达的正确性,选取转录组基因文库内与生长代谢过程中氮代谢相关的谷氨酰胺合成酶(glutamine synthetase,GS)、天冬酰胺合成酶(asparagine synthetase,ASNS)、谷氨酸脱氢酶(glutamate dehydrogenase,GDH)、精氨酸酶(arginase,Argase);碳代谢相关的蔗糖合成酶(sucrose synthase,SuSy)、可溶性淀粉合成酶(soluble starch synthase,SSS)、ADP-葡萄糖焦磷酸化酶(ADP-glucose pyrophosphorylase,AGPase)、淀粉酶(amylase,AMS)和与能量代谢相关的丙酮酸激酶(pyruvate kinase,PK)、己糖激酶(hexokinase,HK)、丙酮酸脱氢酶(pyruvate dehydrogenase,PDH)、苹果酸脱氢酶(malate dehydrogenase,MDH)、异柠檬酸脱氢酶(isocitrate dehydrogenase,IDH)、ATP 酶(adenosine triphosphatase,ATPase)14个具有代表性的基因,用Primer 6 设计引物,以β-actin作为内参基因进行qRT-PCR 相对表达分析[16],根据其cDNA 片段设计特异性引物(表1)。提取RNA后,用HiScript II 1stStrand cDNA Synthesis Kit 试剂盒进行反转录。将反转录产物稀释10 倍,用ChamQTM Universal SYBR ® qPCR Master Mix 做荧光定量。荧光定量在20 μL 体系含有10 μL 2×ChamQTM Universal SYBR qPCR Master Mix,2 μL cDNA 模板和0.4 μL 每种基因特异性引物的反应混合物中进行。最后使用CFX96TMReal-Time System 及其相对定量软件进行2次生物重复3次技术重复。反应参数为95 ℃、30 s,并以95 ℃、10 s,58 ℃、30 s 循环40 次。将cDNA 文库分别标准化为参考基因β-actin。通过2-ΔΔCt方法计算基因相对表达量。
3 结果与分析
3.1 测序数据组装结果
3.1.1 数据过滤统计表 测序后在G 样本中平均获得28.36 Mb 的高质量clean reads,占raw reads 的97.92%,在GA-G 样本平均获得66.27 Mb 的高质量的clean reads,占raw reads的97.84%。4个样本clean reads Q30均大于92%,平均GC 含量为47.28%(表2)。对片段进行拼接得72 244 个Unigene 转录物。
3.1.2 组装质量统计 对片段进行拼接得到72 244个Unigene 转录物。组装结果质量评估可从N50数值来评估。将所有Unigene 从长到短排序,并依次累加长度。当累加片段长度达到总片段长度(所有Unigene 的长度)的50%时,对应那个片段的长度和数量,即为Unigene N50长度和数量。Unigene N50越长,数量越少,说明组装质量越好。在本实验中,由组装结果得N50 长度为1733 bp,其中Unigene 序列最长为15 844 bp,最短为201 bp,平均长度为916 bp,长度在1000 bp 以上的Unigene有18 698 个,占总数的25.88%(表3)。

表1 qRT-PCR 扩增特异引物Table 1 Specific primers of qRT-PCR amplification
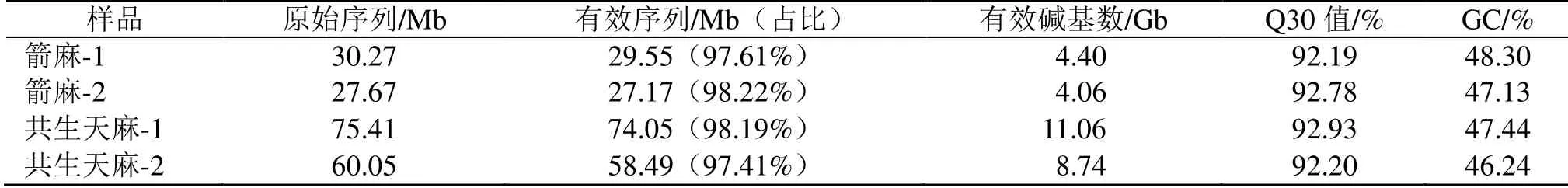
表2 数据过滤统计表Table 2 Data filter statistics table
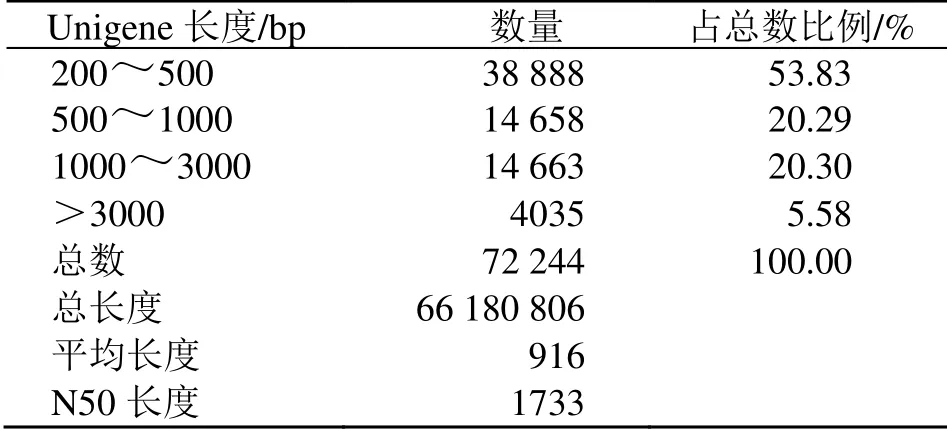
表3 Unigene 长度分布统计Table 3 Length statistics of Unigene distribution
3.1.3 转录物功能注释及分类 为了解转录物Unigene 序列信息,将组装好的72 244 个Unigene序列比对到Swissprot、Nr、KEGG 和KOG 4 个数据库中,有26 312 个Unigenes 获得注释信息,所占比例为36.42%。其中Unigenes 注释成功最多的为Nr 和Swissprot 数据库,分别为26 217 和17 886个。注释在KOG 和KEGG 数据库中的Unigenes 个数分别为16 625 和9 712(表4)。
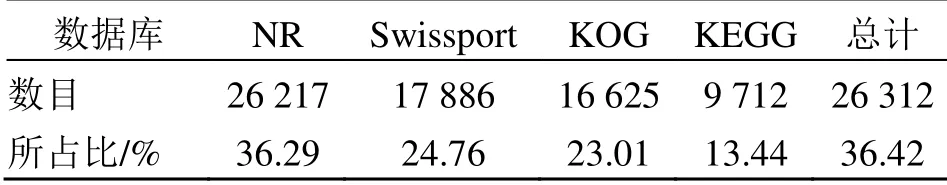
表4 Unigene 功能注释结果统计Table 4 Unigene functional annotation statistical results
3.2 基因差异表达分析
3.2.1 主成分分析( principal component analysis,PCA) 在RNA 组学研究中利用PCA,将样本所包含的上万个维度的信息(上万个基因的表达量),降维为数个维度的综合指标(主成分),以便于进行样本间的比较,同时保证原始数据中包含信息尽可能多地被保留[18]。使用R 语言包(http://www.r-project.org/)对共生天麻和箭麻所有样品进行PCA,如图1 所示。根据共生天麻和箭麻在第1 主成分(PC1)和第2 主成分(PC2)2 个综合指标中的数值大小,做二维坐标图。从图1 可以看出共生天麻-1 和共生天麻-2 分布在右侧,箭麻-1 和箭麻-2 分布在左侧,说明2 种样品具有一定的差异性。PCA 分析中PC1(95.1%)和PC2(4.2%)对样品中所有基因表达量总体方差贡献率为99.3%,表明通过PCA 分析可以很好地区分箭麻和共生天麻。
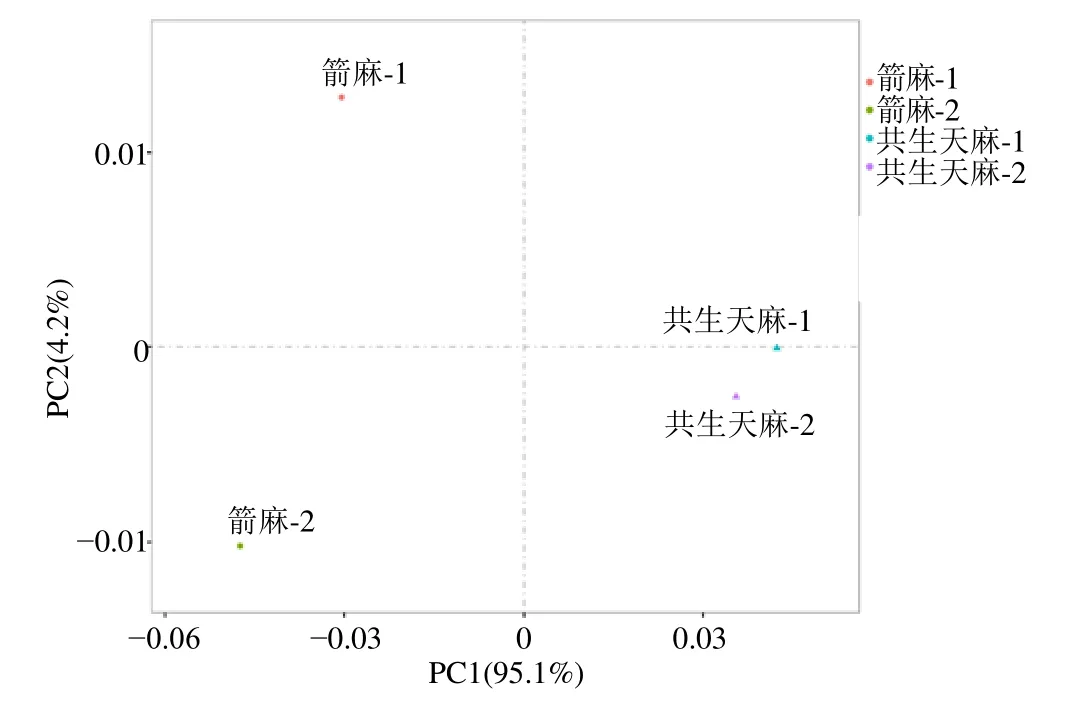
图1 共生天麻和箭麻样品的PCAFig.1 Principal component analysis of symbiotic G.elata and mature tuber
3.2.2 分组间差异基因火山图 比较箭麻与共生天麻的测序结果,以FDR<0.05 且|log2FC|>1 为判断标准,检测到共有12 498 条基因发生显著差异表达,其中上调表达基因数为9 000,占差异表达基因个数的72.01%;下调表达基因数为3 498占差异表达基因数的27.99%。差异表达直观展示火山图见图2。
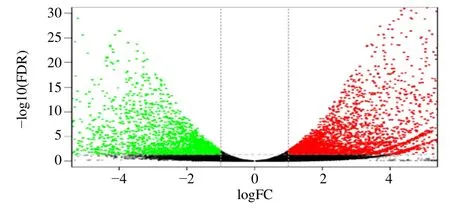
图2 差异表达基因火山图Fig.2 Differential expressed gene volcanic map
3.2.3 差异表达基因的GO 分类 如表5 所示,有10 473 条差异表达基因成功进行了GO 分类。在GO 分类的3 个大类中有4723 条差异表达基因与生物学过程有关,占比为45.10%,其中富集最多的是代谢过程亚类,占比对到该功能类别总数的23.65%;有2508 条差异表达基因与分子功能相关,占比23.95%,其中富集最多的是催化活性亚类,共比对到该功能类别总数46.93%;与细胞成分有关的差异表达基因有3242 条,占比30.96%,富集最多的是细胞和细胞成分亚类,共比对到该功能类别总数的46.33%。
3.2.4 差异表达基因的KEGG 功能分析 箭麻与共生天麻样本间的差异基因进行通路富集分析发现,有803 个差异基因比对到20 个KEGG 通路中。有较多基因归类为苯丙素生物合成(phenylpropanroid biosynthesis)、淀粉与蔗糖代谢(starch and sucrose metabolism)、植物激素信号传导(plant hormone signal transduction)、碳代谢(carbon metabolism)、糖酵解/葡萄糖酵素(glycolysis/gluconeogenesis)和氮代谢(nitrogen metabolism)这几类相关的通路之中,包含了生长过程中氮代谢、碳代谢和能量代谢(如糖酵解)等途径,说明蜜环菌侵入天麻与天麻共生长出箭麻过程中,箭麻与共生天麻生长代谢过程中相关酶基因差异表达,两者的代谢强弱有很大差别(表6)。

表5 箭麻和共生天麻差异表达基因GO 注释Table 5 GO annotation of differentially expressed genes in mature tuber and symbiotic G.elata.

表6 箭麻和共生天麻差异基因的显著富集的代谢途径Table 6 Significant enrichment pathway of differentially expressed genes in mature tuber and symbiotic G.elata.
3.2.5 差异表达基因的KOG 功能注释 在实验中共有26 980 个DEGs 注释到KOG 的25 个分类中,其中7796 个Unigenes 注释为一般功能预测(general functional predictio only,R),占比28.90%;2782个Unigenes 注释为翻译后修饰、蛋白质分解及伴侣分子( posttranslational modification,protein turnover,chaperenes,O),占比10.31%;2295 个Unigenes 注释为信号转导(signal transduction mechanism,T),占比8.51%;1519 个Unigenes注释为RNA 的加工和修饰(RNA processing and modification,A),占比5.63%;774 个Unigenes注释为能量的产生和转化(energy production and conversion,C),占比2.87%;816 个Unigenes 注释为碳水化合物转运与代谢(carbohydrate transport and metabolism,G),占比3.02%,628 个Unigenes注释为次生代谢产物的合成、转运和代谢(secondary metabolites biosynthesis,transport and catabolism,Q),占比2.32%(表7)。
3.2.6 箭麻和共生天麻中生长代谢关键差异基因表达分析 根据差异基因GO 分类、KEGG 功能分析和KOG 功能注释,对箭麻和共生天麻生长代谢过程的相关基因进行筛选。在箭麻和共生天麻转录组测序数据注释的结果中筛选到与氮代谢、碳代谢和能量代谢相关酶的基因序列。在氮代谢途径中,选取了差异表达的基因GS、ASNS、GDH、Argase;在碳代谢中选取了差异表达的基因SuSy、SSS、AGPase、AMS;在能量代谢中选取了差异表达的基因PK、HK、PDH、MDH、IDH、ATPase(表8)。从这些基因的表达趋势来看,与合成氮,碳和能量的酶基因大部分呈上调,除分解能量的酶基因上调外,分解氮,碳的酶基因呈下调。说明蜜环菌侵染天麻与天麻共生时共生天麻各种代谢旺盛,合成各种有机物和能量物质。
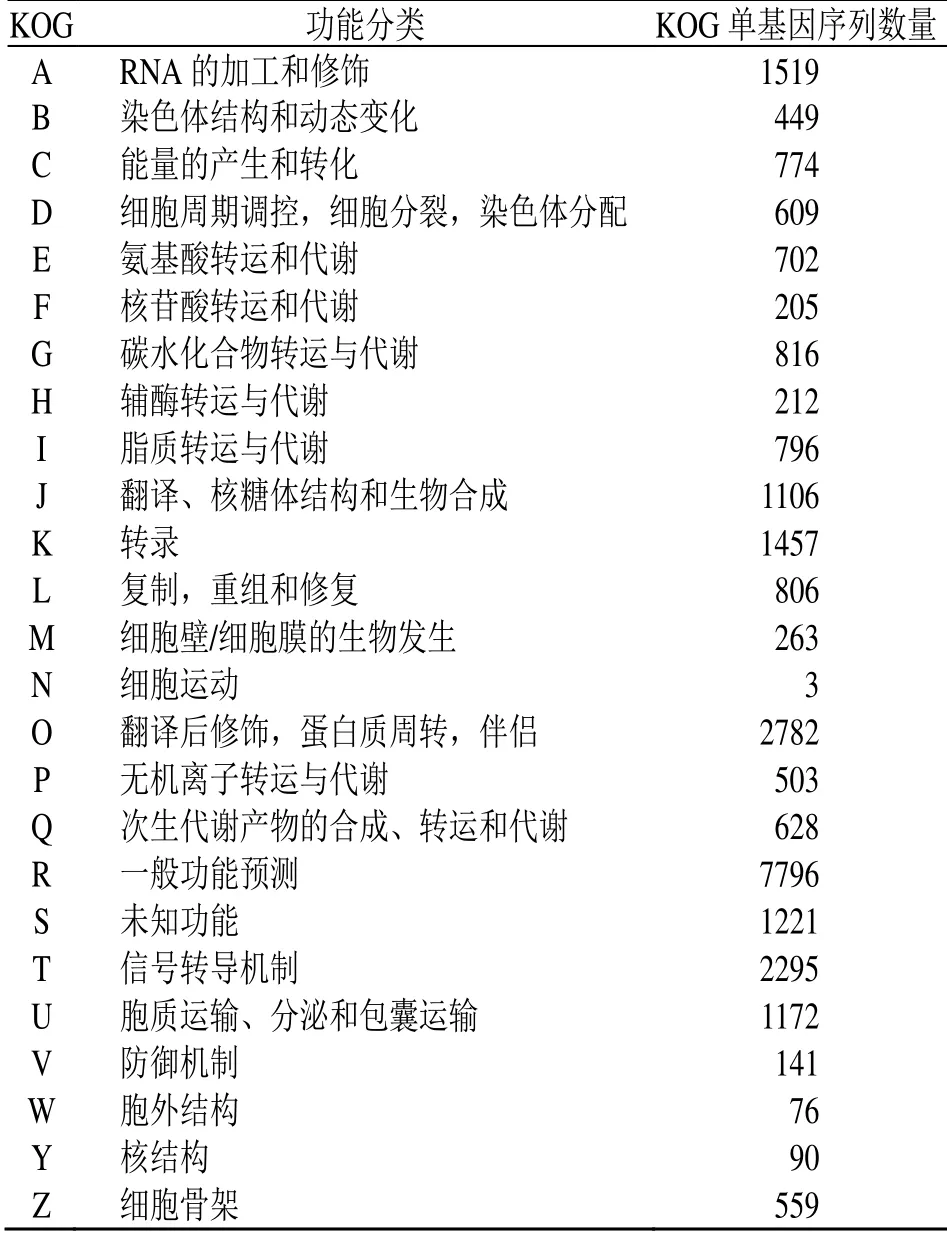
表7 差异表达基因的KOG 功能注释Table 7 DEGs KOG annotation
3.2.7 箭麻和共生天麻中与各种代谢相关基因相对表达水平qRT-PCR 分析 选取的14 个与生长代谢过程相关关键差异酶基因的相对定量qRT-PCR分析结果见图3。由2-ΔΔCt值计算箭麻和共生天麻中的相对表达量,统计分析结果显示箭麻和共生天麻的这些基因表达水平与转录组分析结果一致。即与箭麻相比,共生天麻中合生碳、氮,能量物质的显著差异酶基因呈上调表达,除分解ATP的ATPase基因呈上调表达外,分解氮、碳的GDH、Argase和AMS基因呈下调表达。因此,本课题组认为在转录组分析中用RPKM(Reads Per Kilobase per Million mapped reads,每百万reads 中来自于某基因每千碱基长度的reads 数)法推断的基因表达水平是可靠的。
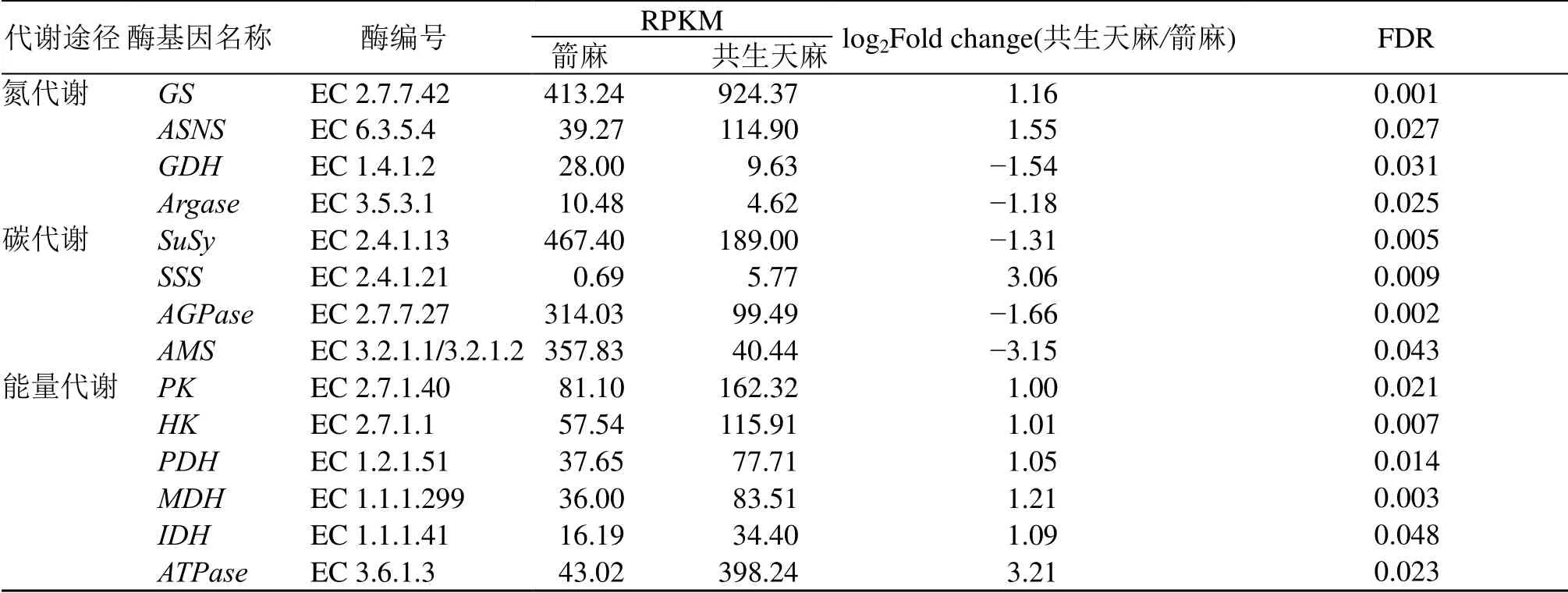
表8 箭麻和共生天麻中与生长代谢相关关键差异基因的表达情况Table 8 Expression of key differentially expressed genes in growth and metabolism of mature tuber and symbiotic G.elata.
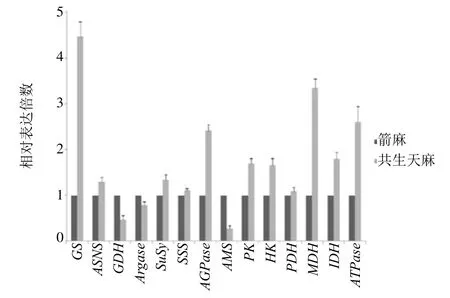
图3 箭麻和共生天麻中生长代谢相关基因相对表达水平分析Fig.3 Analysis of relative expression level of related genes in growth and metabolism in symbiotic G.elata and mature tuber
4 讨论
4.1 共生天麻和箭麻的转录组测序分析
本研究利用Illumina HiseqTM2000 测序技术箭麻和共生天麻进行测序,箭麻和共生天麻分别获得4.23 Gb 和9.90 Gb 有效数。通过数据组装获得72 244 条Unigenes,N50长度为1773 bp,平均长度为916 bp 的非冗余Unigene,且Q30值均大于92%,说明组装数据良好可信。同时将组装好的72 244 个Unigene 序列比对到Swissprot、Nr、KEGG和KOG 4 个数据库中有26 312 个Unigene 获得注释信息,所占比例为36.42%。将箭麻和共生天麻进行PCA,发现箭麻和共生天麻在一定程度上有所差异,通过PCA 可以很好地区分箭麻和共生天麻。在FDR<0.05 和|log2FC|>1 的条件下,比较两组测序数据的结果,发现12 498 条DEGs,上调表达9000条,下调表达3498条。GO数据库将10 473条差异表达基因分为生物学过程、分子功能和细胞组成3 大类,其中代谢过程、细胞过程催化活性等功能小类富集较多。箭麻与共生天麻样本中有803 个DEGs 比对到20 个KEGG 通路中,与箭麻和共生天麻的生长代谢过程相关的淀粉与蔗糖代谢、碳代谢、糖酵解/葡萄糖酵素等表现出显著差异。将DEGs 注释到KOG 的25 个分类中,含有能量的产生和转化,碳水化合物转运与代谢和次生代谢产物的合成、转运和代谢功能分类。根据DEGs 的GO 分类,KEGG 功能分析和KOG 功能注释筛选出箭麻和共生天麻生长代谢(氮代谢、碳代谢和能量代谢)过程中的关键差异酶基因。qRT-PCR分析结果与转录组数据结果基本一致。在差异表达的基因中,筛选到与天麻氮、碳和能量代谢相关基因,合成氮和碳营养物质的差异基因表达上调,分解氮碳营养物质的差异基因表达下调,能量相关的差异基因大多数表达上调。与箭麻相比较,共生天麻有利于氮、碳营养物质和能量物质合成。
4.2 共生天麻和箭麻中生长代谢过程相关关键差异基因分析
氮、碳和能量的积累在植物生长过程中至关重要,因此选取了氮代谢,碳代谢和能量代谢过程中的关键酶基因。氮素同化在植物生长和发育过程中是一个十分重要的生理过程。植物从外界吸收的无机铵态氮和硝态氮必须先同化为谷氨酰胺和谷氨酸等有机物才能被植物利用。氨同化过程中谷氨酰胺合成酶是一个关键酶,它在ATP 供能条件下催化NH4+同化为谷氨酰胺,然后在谷氨酸合酶作用下将其转化生成谷氨酸。许多研究表明正常条件下高等植物氨同化的主要途径是谷氨酰胺合成酶和谷氨酸合酶构成的循环反应途径[19]。谷氨酸脱氢酶参与催化谷氨酸分解为α-酮戊二酸和铵离子及其逆反应合成谷氨酸的可逆反应。天冬酰胺和精氨酸是植物体内N/C 比值较高的氨基酸,是植物氮代谢过程中有机氮运输和储存的主要形式。近年来进一步研究表明,苹果树在越冬期间其体内可溶性和蛋白态氨基酸中,精氨酸含量均很高,这说明苹果树中的氮素在越冬期间主要以精氨酸形式贮藏[20-22]。qRT-PCR 分析表明共生天麻GDH和ASNS基因都明显上调,分解谷氨酸的GDH基因和分解精氨酸的Argase基因都呈下调,这些基因不同表达水平有利于共生天麻积累便于在植物体内储存运输的谷氨酰胺,谷氨酸、天冬酰胺和精氨酸,为共生天麻和箭麻的基础代谢和生长代谢提供氮素营养。
淀粉是高等植物在生长、发育和繁殖过程中重要碳水化合物营养物质的贮藏形式,并且以往研究表明蔗糖主要参与淀粉等的合成。植物蔗糖代谢过程的关键酶是蔗糖合酶,它催化蔗糖的合成和分解,但研究表明它主要催化蔗糖分解为淀粉合成所需的葡萄糖和果糖[23]。并有研究表明蔗糖合酶作为关键代谢酶在马铃薯块茎淀粉合成中为其提供前体物质二磷酸尿核苷葡萄糖(uridine diphosphate glucose,UDPG)[24],马铃薯中过量表达该酶可引起二磷酸腺苷葡萄糖(adenosine diphosphate glucose,ADPG)和UDPG 的含量增加,淀粉产量增加了近1 倍,同时马铃薯干质量较野生型也有所增加[25]。植物合成淀粉的另一个限速酶是AGPase,研究表明AGPase 是淀粉生物合成过程中的关键酶,也是淀粉合成的限速酶,抑制ADP-葡萄糖焦磷酸化酶活性将会部分或全部终止淀粉的合成[26-27]。可溶性淀粉合成酶也是淀粉合成过程中的关键酶。它以腺苷二磷酸葡萄糖为葡萄糖的供体,催化形成淀粉颗粒。淀粉酶是能将淀粉分子中的糖苷键水解获得葡萄糖和寡糖等产物的一类酶。转录组分析和qRT-PCR 结果都显示相较于箭麻,共生天麻中SuSy、AGPase和SSS基因均上调,AMS基因下调,这些基因的差异表达有利于共生天麻淀粉的合成和累积,供共生天麻和箭麻的生长发育需要。
糖酵解和柠檬酸循环是产生能量的主要途径。丙酮酸激酶和己糖激酶是糖酵解途径的限速酶[28]。一分子葡萄糖通过糖酵解途径分解为2 分子丙酮酸可以产生2 分子ATP。苹果酸脱氢酶和异柠檬酸脱氢酶是柠檬酸循环中主要限速酶[28]。一分子丙酮酸通过柠檬酸循环可以产生12.5 分子ATP。通过糖酵解和柠檬酸循环分解一分子葡萄糖可产生32 分子ATP。ATP 酶能催化ATP 水解为ADP 和磷酸根离子,并释放能量的反应。基于转录组数据和 qRT-PCR 分析,共生区天麻HK、PDH、IDH、MDH基因都表达上调,说明共生区天麻糖酵解和柠檬酸循环都处于快速代谢产生较多的ATP 储存能量。同时ATP基因也呈上调表达,它催化ATP基因分解释放较多的能量以满足共生天麻与蜜环菌共生所需要的能量和箭麻生长代谢所需要的能量。
5 结论
近年来从分子水平上对天麻及天麻与蜜环菌共生的研究颇多,最新研究已从生物学角度揭示天麻与蜜环菌共生关系及其形成机制[16]。但是天麻(白麻)与蜜环菌共生后进一步生长出新天麻(箭麻)的生长代谢特征还不清楚。本研究通过转录组比较分析初步阐述共生天麻和箭麻生长代谢特征。转录组分析表明,共生天麻中与氮、碳和能量代谢相关差异基因与箭麻相比较大多数合成氮、碳和能量物质表达上调,分解谷氨酸和精氨酸的谷氨酸脱氢酶和精氨酸酶以及分解淀粉的淀粉酶表达下调,有利于共生区天麻产生较多的氮和碳营养储存物质和较多的能量物质供其进一步生长为箭麻。可以看出白麻与蜜环菌共生状况是否良好对白麻能否进一步生长出箭麻有很大影响。在生产中科学地掌握白麻与蜜环菌共生时适宜的生长环境是白麻可以进一步生长为箭麻的关键,这是天麻栽培过程中需要注意的事项。然而,天麻蜜环菌共生时的最适宜环境仍需要进行下一步的探究。与此同时,箭麻和共生天麻与碳代谢、氮代谢及能量代谢相关的基因中,同一基因家族成员基因时空表达不一致,今后应研究清楚基因家族成员在箭麻和共生天麻中的亚细胞定位和时空表达特性,阐明其功能与作用,为人工栽培天麻提供进一步理论指导。
利益冲突所有作者均声明不存在利益冲突
