阿可拉定纳米混悬剂的处方设计、优化与质量评价
2021-02-03刘媛媛咸阳职业技术学院咸阳72000陕西中医药大学第二附属医院咸阳72000
刘媛媛,贺 朝(.咸阳职业技术学院,咸阳 72000;2.陕西中医药大学第二附属医院,咸阳 72000)
阿可拉定(Ica)又称淫羊藿苷元,是从多年生草本植物淫羊藿中分离的淫羊藿提取物经酶转化得到的一种异戊二烯黄酮类化合物单体[1]。早期研究发现,Ica具有抗炎、抑制骨质疏松、保护神经系统和调节免疫等药理作用[2],近年来研究发现,Ica能够抑制肝癌、肺癌和乳腺癌等肿瘤细胞增殖,诱导其凋亡等药理活性[3]。Ica在水中的溶解度小于1.0 μg·mL-1,导致口服吸收不完全,生物利用度较低,降低了药物的治疗效果[4]。为了改善Ica的溶解度,研究人员将其制备成胶束[5]、磷脂复合物[6]和脂质体[7]等新型给药系统。本研究将Ica制备成纳米混悬剂(Ica-NSs),采用Box-Behnken实验设计优化得到Ica-NSs的最优处方,为Ica的体内药动学研究奠定实验基础。
1 仪器与材料
1.1仪器 LGJ-18N型普通多歧管冷冻干燥机(北京亚星仪科科技发展有限公司);JY98-ⅢN型超声波细胞粉碎机(宁波新芝生物科技股份有限公司);S10-3型数显恒温磁力搅拌器(上海司乐仪器有限公司);RYX-12D 型全自动溶出取样收集系统(北京北研科仪仪器有限责任公司);JSM-IT500型扫描电子显微镜(日本电子株式会社);Malvern Zetasizer Nano型激光纳米粒度仪(英国马尔文公司)。
1.2材料 阿可拉定原料药(上海熹垣生物科技有限公司,质量分数为98.5%,批号20180923);羟丙基纤维素(HPC SSL,日本曹达株式会社);羟丙甲纤维素(HPMC E5,亚什兰中国公司);十二烷基硫酸钠(SDS,德国默克密理博公司);聚维酮(PVP K30)和泊洛沙姆188(F68)均购于上海巴斯夫辅料公司;吐温80(T-80,日本油脂公司)。
2 方法与结果
2.1Ica-NSs的制备 采用反溶剂沉淀-超声法[8-9]制备Ica-NSs。①配制药物乙醇溶液:称取处方量Ica加入到2 mL乙醇中溶解,经0.45 μm微孔滤膜过滤,除去不溶性固体杂质,得到药物的有机相溶液,备用;②配制反溶剂:按照实验设计称取处方量稳定剂(HPC SSL、PVP K30和HPMC E5)和表面活性剂(SDS、F68和T-80),溶解到一定体积的蒸馏水中,经0.8 μm微孔滤膜过滤,得到反溶剂,备用;③Ica-NSs的制备:设置磁力搅拌速度为1 500 r·min-1,通过带有7号针头的注射器将含药物的有机相溶液滴加到反溶剂中,快速析出药物晶体,持续搅拌10 min,在45 ℃条件下真空除去乙醇;将上述悬浊液经探头超声(超声2 s,间隔2 s,超声功率为380 W)处理5 min,即得到Ica-NSs。
取新制备的Ica-NSs 5 mL,加入15 mL西林瓶中,加入质量浓度为100 g·L-1的甘露醇,溶解,置于冷冻干燥机中冷冻干燥,预冻温度为-40 ℃,一次干燥温度为-5 ℃,真空度为30 Pa,干燥时间为10 h,解析干燥温度为25 ℃,真空度为5 Pa,干燥时间为6 h,干燥结束后加塞密封保存。
2.2含量测定方法
2.2.1色谱条件 ZORBAX SB-C18色谱柱(150 mm×4.6 mm,5 μm);流动相:10 g·L-1磷酸-甲醇(20∶80);检测波长:272 nm;流速:1.0 mL·min-1;进样量:10 μL;柱温:40 ℃。
2.2.2线性考察 精密称取Ica对照品10 mg,置于20 mL量瓶中,加入甲醇5 mL溶解,再加入流动相定容,得到对照品储备液,取上述储备液加入流动相逐步稀释成药物质量浓度分别为50.0,10.0,5.0,1.0,0.5 μg·mL-1的溶液,进样检测。以药物质量浓度为横坐标,峰面积为纵坐标,进行线性回归,得到方程为A=49 325.6C+2 902.6(r=0.999 9),说明Ica质量浓度在0.5~50.0 μg·mL-1范围内线性关系良好。取低、中、高(0.5,5.0,50.0 μg·mL-1)3个质量浓度Ica对照品溶液,分别进行日内精密度与日间精密度考察,结果显示,低、中和高3个质量浓度的Ica对照品溶液的日内精密度RSD值分别为0.9%,1.0%,0.5%(n=6),日间精密度RSD值分别为1.2%,0.8%,1.1%(n=6),表明方法精密度良好。
2.3Ica-NSs处方研究
2.3.1稳定剂种类筛选 文献报道[10],稳定剂的种类和质量浓度会对纳米混悬剂的粒径分布产生显著影响,因此本研究需要对稳定剂的种类进行筛选。高分子聚合物稳定剂选择PVP K30、HPC SSL和HPMC E5,表面活性剂选择SDS、F68和T-80,将二者联合应用考察其对Ica-NSs制剂性质的影响,结果见表1。
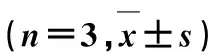
表1 稳定剂筛选结果
由表1可知,以PVP K30和SDS联合使用作为稳定剂制备的Ica-NSs粒径分布较小,Zeta电位绝对值较大,放置12 h未发生聚集和沉降现象。因此本研究选择PVP K30和SDS作为Ica-NSs的稳定剂,并对二者的质量浓度做进一步的研究。
2.3.2稳定剂质量浓度筛选 固定Ica质量浓度为10 mg·mL-1,分别考察PVP K30质量浓度为2,6 mg·mL-1,SDS质量浓度为0.4,1.0 mg·mL-1,制备Ica-NSs,测定粒径分布、PDI及Zeta电位,筛选稳定剂的质量浓度,结果见表2。
表2 稳定剂质量浓度筛选结果
研究结果表明,稳定剂质量浓度对Ica-NSs的粒径分布影响较大,当体系中PVP K30质量浓度较低时,制备的Ica-NSs粒径相对较小,而PVP K30质量浓度较高时,溶液黏度增大,促使Ica-NSs粒子之间相互黏附聚集,粒径变大,因此需要对稳定剂质量浓度进一步优化。
2.3.3Ica质量浓度筛选 固定PVP K30质量浓度为2 mg·mL-1、SDS质量浓度为0.4 mg·mL-1,选择Ica质量浓度分别为10,30,50 mg·mL-1制备Ica-NSs,测定粒径分布、PDI及Zeta电位,筛选Ica质量浓度,结果见表3。
表3 Ica质量浓度筛选结果
研究结果表明,Ica质量浓度由10 mg·mL-1增加到50 mg·mL-1时,制备的Ica-NSs粒径从(278.2±26.7) nm增加到(494.3±29.3) nm,说明粒径随着Ica质量浓度的增加而增大,因此需要对Ica质量浓度进一步优化。
2.4Ica-NSs处方优化
2.4.1Box-Behnken实验设计 通过前期处方筛选结果可知,粒径大小对Ica-NSs的稳定性起到关键作用,因此本研究以纳米混悬剂的粒径分布(Y)作为评价指标,以Ica质量浓度(X1)、PVP K30质量浓度(X2)和SDS质量浓度(X3)作为变量因素,自变量水平见表4。利用Box-Behnken实验设计法对Ica-NSs的处方组成进行优化,实验设计及结果见表5。
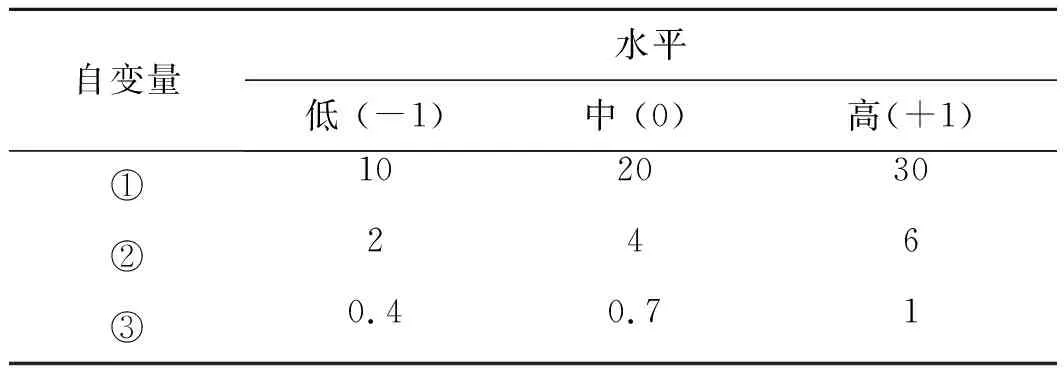
表4 Box-Behnken实验设计自变量与水平

表5 Box-Behnken实验设计及结果
2.4.2模型的建立及显著性检验 通过Box-Behnken实验设计软件对表5数据进行分析处理,以评估Ica、PVP K30和SDS的质量浓度与Ica-NSs粒径分布之间的相关性。方差分析结果见表6。由表6可知,模型Y的P值均为0.000 8,小于0.05,说明建立的模型显著;而失拟项P值为0.389 1,大于0.05,说明实测值和预测值之间差异无统计学意义,模型拟合可信度较高。建立的模型拟合方程为:
Y=822.54-44.98X1+9.93X2-287.64X3+0.54X1X2+2.71X1X3+34.75X2X3+0.97X12-0.03X22+115.79X32(R2=0.982 0)。
结果表明,Ica、PVP K30和SDS的质量浓度对Ica-NSs的粒径分布影响显著(P<0.05)。通过效应面图更能直观地分析自变量与因变量之间的关系。效应面图见图1。
表6 方差分析结果

图1 Ica质量浓度(X1)、PVP K30质量浓度(X2)和SDS质量浓度(X3)对Ica-NSs的粒径分布(Y)的效应面图
由图1可知,当PVP K30质量浓度恒定时,随着Ica质量浓度的增加,Ica-NSs的粒径呈先减小后增大趋势;当Ica质量浓度恒定时,随着PVP K30质量浓度的增加,Ica-NSs的粒径呈增大趋势;当PVP K30质量浓度恒定时,随着SDS质量浓度的增加,Ica-NSs的粒径呈增大趋势。
本研究要求制备的Ica-NSs具有最小化的粒径分布,经软件优化得到最佳处方组成为:Ica、PVP K30和SDS质量浓度分别为20.0,3.0,0.7 mg·mL-1,预测得到Ica-NSs的粒径分布为340.6 nm。根据最佳处方制备Ica-NSs,测定粒径分布为(335.2±18.5) nm,实验测定值与预测值接近,模型预测准确性较高。
2.5Ica-NSs及冻干粉末表征
2.5.1扫描电镜观察 取少量Ica-NSs,用蒸馏水稀释,取1滴加到锡箔纸上,挥干水分,黏附至电胶布上,喷金;另取冻干后的Ica-NSs固体粉末,黏附至电胶布上,喷金。在扫描电镜下分别观察2份样品的微观结构,并拍摄电镜照片,见图2。

图2 Ica-NSs(A)及其Ica-NSs冻干粉末(B)扫描电镜图
在扫描电镜下可观察到冻干前Ica-NSs粒径较小,大部分粒径分布在200~400 nm,且颗粒呈不规则状态;Ica-NSs冻干粉末呈多孔纤维状结构,可以看到Ica-NSs沉积在表面。
2.5.2粒径分布测定 取Ica-NSs及该批Ica-NSs制备的冻干粉末,加入蒸馏水稀释,并轻轻振摇,分散成透明状溶液,取上述样品,采用Malvern Zetasizer Nano型激光纳米粒度仪测定粒径分布、PDI及Zeta电位,结果见表7。
由表7可知,Ica-NSs冻干前后的粒径分布、PDI和Zeta电位未发生显著变化,说明冷冻干燥没有促使其聚集。
表7 Ica-NSs冻干前后的理化性质
2.6溶出度对比研究 比较Ica原料药、Ica-NSs及Ica-NSs冻干粉末的体外药物溶出度,溶出度测定参数为:介质为pH6.8磷酸盐缓冲液(加入10 mg·mL-1T-80),体积为500 mL,搅拌桨转速为50 r·min-1。分别取Ica原料药,Ica-NSs及Ica-NSs冻干粉末,加入到溶出杯中,依次在5,10,15,30,45,60 min取出5 mL介质溶液,过滤,稀释至适当质量浓度,检测药物含量,计算药物溶出量,绘制时间-溶出曲线。见图3。

图3 Ica原料药、Ica-NSs及Ica-NSs冻干粉末的体外溶出曲线 (n=6)
实验结果表明,Ica-NSs在10 min内药物溶出度基本达到100%,Ica-NSs冻干粉末在5 min时溶出度较纳米混悬剂稍慢,到10 min时药物溶出度与纳米混悬剂基本一致,Ica原料药溶出非常缓慢,在60 min溶出度仅为50%左右。说明将Ica制备成纳米混悬剂可显著提高药物的溶出度。
2.7稳定性研究 取Ica-NSs冻干粉末密封于西林瓶中,放置在加速实验(40 ℃、相对湿度为75%)条件下考察其稳定性,在设定时间间隔取样,测定其粒径分布、PDI、Zeta电位、含量以及10 min药物溶出度,结果见表8。稳定性实验结果表明,Ica-NSs冻干粉末在加速条件放置3个月后粒径分布、PDI、Zeta电位、含量以及10 min药物溶出度几乎无变化,说明其稳定性良好。
表8 加速实验稳定性结果
3 讨论
纳米混悬剂是以少量高分子聚合物和/或表面活性剂作为稳定剂,通过机械性超微粉碎技术或者控制结晶析出条件将药物制备成粒径小于1 000 nm且具有良好的物理稳定性的胶体分散体[11],由于纳米混悬剂粒径较小,将药物制备成纳米混悬剂后能够显著增加药物的溶解度和生物利用度[12-13]。然而,纳米混悬剂比表面积巨增,热力学体系处于不稳定状态,因此会自发地通过相互聚集以减小比表面积,最终导致纳米混悬剂出现聚集、团聚、沉降和晶型转变等现象,因此需要在处方中加入稳定剂阻止纳米混悬剂的聚集或团聚,以增加体系的稳定性,目前常用的稳定剂分为高分子聚合物类和表面活性剂2种类型[14-16]。本研究通过实验筛选表明,以PVP K30和SDS联合使用作为稳定剂制备的Ica-NSs粒径分布较小,Zeta电位绝对值较大,放置12 h体系未发生聚集和沉降现象,这是由于PVP K30作为聚合物高分子能有效地吸附在纳米混悬剂表面,产生空间位阻作用,另外SDS吸附在纳米混悬剂表面增加了微粒之间的静电作用,进一步阻碍纳米混悬剂聚集,通过PVP K30和SDS在纳米混悬剂表面形成紧密的聚合物/表面活性剂二元保护层,提高了体系的稳定性。
目前文献报道,对于纳米混悬剂的处方筛选和优化可以选择正交实验设计法[17]、中心复合实验设计法[18]和Box-Behnken实验设计法[19]等,每种实验设计各有特点,正交实验设计只能给出所筛选实验的最佳组合,而Box-Behnken实验设计法可以克服正交实验设计存在的弊端,可以在整个区域内提供最佳实验组合,该设计使用方便,预测性良好,是解决实际问题的有效手段[20]。本实验采用了Box-Behnken实验设计法优化了Ica-NSs的处方,实测值与预测值接近,预测准确性较高。
