超高效液相色谱-串联质谱法检测牛组织中常山酮残留量
2021-01-28
(漯河食品职业学院,河南 漯河 462300)
常山酮(halofuginone)属于人工合成的一种广谱抗球虫药,是一种从植物常山中分离出来的常山碱的卤代衍生物[1],其氢溴酸盐治疗效果较好、副作用较少,且不易产生交叉耐药性,被广泛用来防治畜禽的球虫病[2-3]。常山酮主要在肝脏进行代谢,在组织中降解较快[4],孙晓娟等[5]、吴宁鹏等[6]研究发现常山酮的休药期为5 d,为此,在用药过程中,应严格执行其休药期,确保动物性食品的安全[7]。目前,常山酮在畜禽养殖上已大量使用,因此,建立可靠的牛组织中常山酮残留量的检测方法,科学推测其休药期,保证食品安全显得尤为重要。
目前,常山酮残留量的检测方法主要包括液相色谱法[8-11]、酶联免疫法[12-13]和液相色谱-串联质谱法[14-16]等。在动物性食品中,常山酮残留量检测的相关国家标准方法[17-18]均为液相色谱法。依据GB 29693—2013《食品安全国家标准动物性食品中常山酮残留量的测定高效液相色谱法》和SN 0643—1997《出口肉及肉制品中溴氯常山酮残留量检验方法》,动物性食品中的常山酮残留量的检测定量限均为50μg/kg,而GB31650—2019《食品安全国家标准食品中兽药最大残留限量》[19]中要求牛组织中常山酮残留限量为10 μg/kg~30 μg/kg,因此,采用GB 29693—2013和SN 0643—1997对牛组织中常山酮的残留量进行检测,其定量限均无法达到牛组织中常山酮残留限量的标准要求,本试验对牛组织中常山酮残留量的前处理方法进行了优化,采用液相色谱-串联质谱法进行检测,以期解决牛组织样品中常山酮残留限量低,国家相关检测标准定量限达不到残留限量标准要求的问题,将其用于牛组织、鸡组织等不同基质样品中常山酮残留量的快速定性和定量。
1 材料与方法
1.1 主要仪器
Xevo TQ MS超高效液相色谱-串联质谱仪、JLSPE-12B型固相萃取装置:Waters公司;BSA224S型电子天平、ML802E型电子天平:Mettler Toledo公司;GL-21M型离心机:湘仪离心机有限公司;VX-III型多管平行振荡器:Targin公司;RE-201D旋转蒸发装置:Eyela公司;5200DE型数控超声波清洗器:昆山市超声仪器有限公司。
1.2 试剂和材料
除非另有说明,分析中仅使用分析纯的试剂,水为符合GB/T 6682—2008《分析实验室用水规格和试验方法》规定的一级水;氢溴酸常山酮对照品(含量≥99.6%):中国标准物质中心;牛肉、牛肝样品:市售;Oasis HLB 固相萃取柱(60 mg,3 mL):Waters公司;尼龙滤膜(0.22 μm):天津津腾实验室设备有限公司;乙酸铵、冰乙酸(均为优级纯):上海国药集团;甲醇、乙酸乙酯、甲酸铵、甲酸(均为色谱纯)、乙腈(质谱纯):Fisher公司。
1.3 主要溶液配制
1.3.1 0.25 mol/L乙酸铵缓冲液
称取19.27 g乙酸铵用水溶解于1 000 mL容量瓶中,加入30 mL冰乙酸,用水定容至1 000 mL,用冰乙酸调pH值至4.3。
准确量取150 mL乙腈、445 mL水和5 mL冰乙酸,再加入1.73 g乙酸铵,混匀,现配现用。
1.3.3 20 mg/mL胰蛋白酶溶液
准确称取20 g胰蛋白酶,用水溶解并定容至1 000 mL。
1.3.4 200 μg/mL常山酮标准储备液
准确称取氢溴酸常山酮对照品10.0 mg于50 mL棕色容量瓶中,用0.25 mol/L乙酸铵缓冲液溶解并稀释至刻度。2℃~8℃避光保存,有效期6个月。
1.3.5 2.0 μg/mL常山酮标准储备液
准确吸取200 μg/mL常山酮标准储备液1 mL于100 mL棕色容量瓶中,用水溶解并稀释至刻度。2℃~8℃避光保存,有效期3个月。
有时再谈得远一点,就是表姊表妹之类订了婆家,或是什么亲戚的女儿出嫁了。或是什么耳闻的,听说的,新娘子和新姑爷闹别扭之类。
1.4 仪器条件
1.4.1 液相色谱条件
色谱柱:WatersBEH-C18(2.1mm×50mm,1.7μm);流动相:A相为0.1%甲酸水溶液(含5 mmol/L甲酸铵),B相为乙腈;流速:0.45 mL/min;进样量:5 μL;柱温:40℃;流动相及梯度洗脱条件见表1。

表1 常山酮流动相及梯度洗脱条件Table 1 Mobile phase and gradient elution conditions of halofuginone
1.4.2 质谱条件
电离模式:电喷雾正离子(ESI+)模式;离子源温度:150 ℃;毛细管电压:3.2 kV;干燥气流量:900 L/Hr;干燥气温度:450℃;锥孔反吹气流量:40 L/Hr;检测模式:多反应监测(multi-reaction monitoring,MRM)模式,监测条件如表2所示[20]。

表2 常山酮的多反应监测(MRM)条件Table 2 Multi-reaction monitoring(MRM)conditions of halofuginone
1.5 样品处理
1.5.1 样品提取
称取试样(4.00±0.04)g于50 mL离心管中,加入10 mL 20 mg/mL胰蛋白酶溶液,涡动1 min,用10%碳酸钠溶液调pH值至8.0,涡旋混匀后于40℃水浴中酶解3 h。取出,放至室温(20±5)℃,加入10%碳酸钠溶液2 mL,涡动1 min,再加入乙酸乙酯20 mL,于多管平行振荡器上振荡5 min,0℃离心机中放置3 min,0℃环境中2000r/min离心3min,将上清液转入100mL蒸馏瓶中,下层液用乙酸乙酯20 mL重复提取一次,合并两次上清液[17-18]。上清液于40℃旋转蒸发仪上蒸干,残余物中加入0.125 mol/L乙酸铵缓冲溶液10 mL和水饱和正己烷10 mL,于超声波提取器中振荡提取1 min,转入另一50mL离心管中,7000r/min离心5min,收集下层水相,备用。
1.5.2 样品净化
HLB柱依次用 3 mL甲醇、3 mL水和 3 mL 0.125 mol/L乙酸铵缓冲液活化,取全部备用液过柱,用3 mL水淋洗柱子;最后用5 mL甲醇洗脱,收集洗脱液,于40℃旋转蒸发仪上蒸干,用1.0 mL流动相(1.3.2)溶解残余物,混匀,过0.22 μm滤膜,供液相色谱-串联质谱检测。
1.5.3 基质匹配标准曲线的配制
精密吸取2.0 μg/mL的常山酮标准储备液适量,用甲醇溶解并稀释,配制成浓度为 0.5、1、5、10、20、50 ng/mL的系列标准溶液,各取1.0 mL,分别加至经提取、净化步骤处理的空白试样洗脱液中,于40℃旋转蒸发仪上蒸干,用1.0 mL流动相(1.3.2)溶解残余物,混匀,过0.22 μm滤膜,供液相色谱-串联质谱测定。
2 结果与讨论
2.1 样品处理的优化
试样按1.5.1酶解并经乙酸乙酯提取后,若采用0.125 mol/L乙酸铵缓冲液反萃取,在乙酸铵缓冲液中会溶解少量乙酸乙酯,导致样品净化时在上样过程中,少量乙酸乙酯将常山酮部分洗脱,从而影响检测回收率。本试验中,样品酶解并经乙酸乙酯提取后,采用旋蒸至干,然后用0.125 mol/L乙酸铵缓冲液复溶,消除了乙酸乙酯对常山酮的洗脱,从而提高了检测的回收率。
试验表明:采用HLB固相萃取柱净化上样,当乙酸铵缓冲液中不含乙酸乙酯时,常山酮的回收率为90%以上;当乙酸铵缓冲液中含5%乙酸乙酯时,常山酮的回收率降至40%左右;当乙酸铵缓冲液中含10%乙酸乙酯时,常山酮基本无回收。经优化,本试验采用将乙酸乙酯旋蒸至干后,再用0.125 mol/L乙酸铵缓冲液溶解残渣,并用水饱和正己烷除脂肪,以确保净化上样过程中无乙酸乙酯残留,从而大大提高检测回收率。
为减少基质效应,保证检测回收率,本试验采用基质匹配标准曲线进行定量,优化后样品处理方法见1.5。
2.2 液相色谱-串联质谱法仪器条件的优化
2.2.1 质谱条件的优化
在电喷雾离子源正离子(ESI+)模式下,常山酮易得到H+形成较为稳定的[M+H]+的分子离子。本试验采用100 ng/mL常山酮标准溶液进行全扫描和子离子扫描,获得常山酮的母离子扫描图(图1)和特征碎片离子扫描图(图2),并对其毛细管电压、碎裂电压和碰撞能量等条件进行优化,选择离子丰度最强的碎片离子对为定量离子对,离子丰度相对较强的离子对为定性离子对,优化多反应监测条件。
2.2.2 液相条件的优化
本试验采用BEH-C18色谱柱对常山酮进行分离,以乙腈作为流动相中的有机相,对比了水、0.1%甲酸水溶液和0.1%甲酸水溶液(含5 mmol/L甲酸铵)作为流动相中的水相对常山酮的峰型和灵敏度的影响。结果表明,当水相中加入微量甲酸时,常山酮的灵敏度显著提高,而加入微量甲酸铵时,目标物的峰型变得尖锐对称,所以最终选用0.1%甲酸水溶液(含5 mmol/L甲酸铵)作为水相。经优化,最终选用1.4.1中液相条件对常山酮进行洗脱,采用优化后液相条件测定的1 ng/mL常山酮标准溶液的总离子流图和定量离子色谱图如图3所示。
2.3 线性方程和定量限
按1.5.3中的常山酮标准系列配制标准溶液,按1.4中建立的仪器条件进行测定。以测定峰面积Y对含量X(ng/mL)绘制标准曲线,其回归方程为Y=18 283.5X+203.8,相关系数R2=0.998 6。以基质标准溶液3倍信噪比为检出限,10倍信噪比为定量限,得到常山酮的检出限为 0.25 μg/kg,定量限为 0.5 μg/kg。
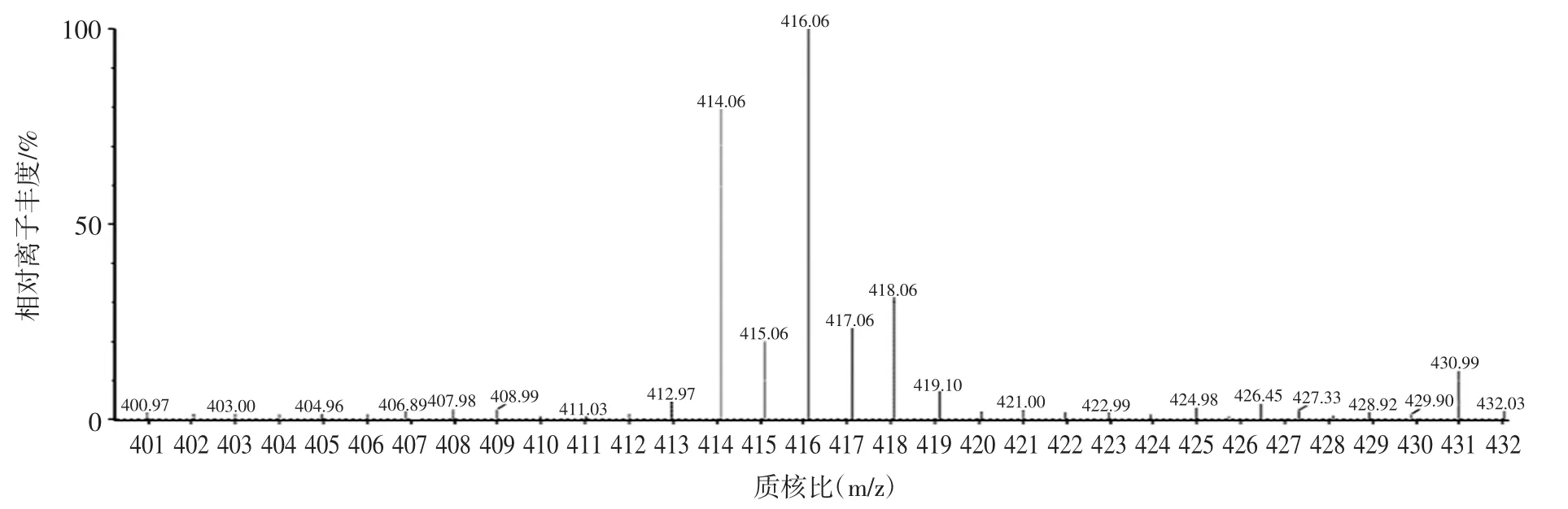
图1 常山酮的母离子扫描图Fig.1 The parent ion scan of halofuginone
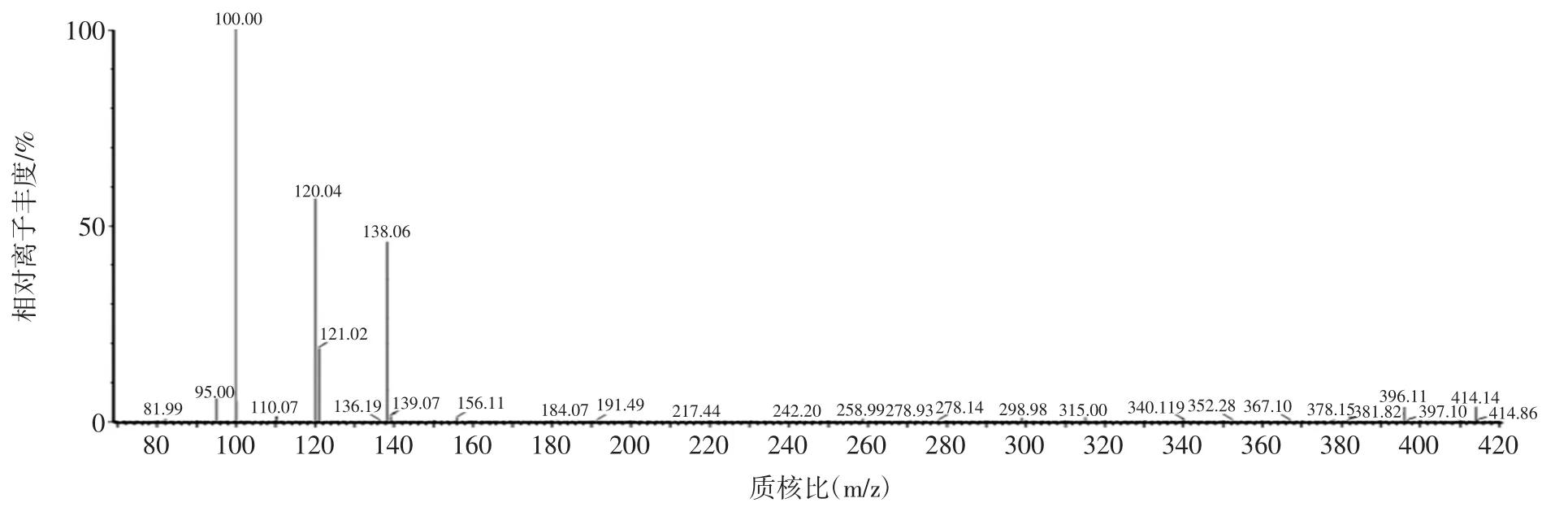
图2 常山酮的特征碎片离子扫描图Fig.2 The fragment ions scan of halofuginone

图3 1 ng/mL常山酮标准溶液的总离子流图和定量离子色谱图Fig.3 The total ion chromatogram and quantitative ion chromatogram of 1 ng/mL halofuginone
2.4 加标回收率和精密度
取处理均匀的牛肉和牛肝样品,按表3设计试验。结果表明,牛组织中常山酮的回收率为73.2%~93.60%,相对标准偏差(relative standard deviation,RSD)为5.91%~7.96%,具体结果如表3所示。
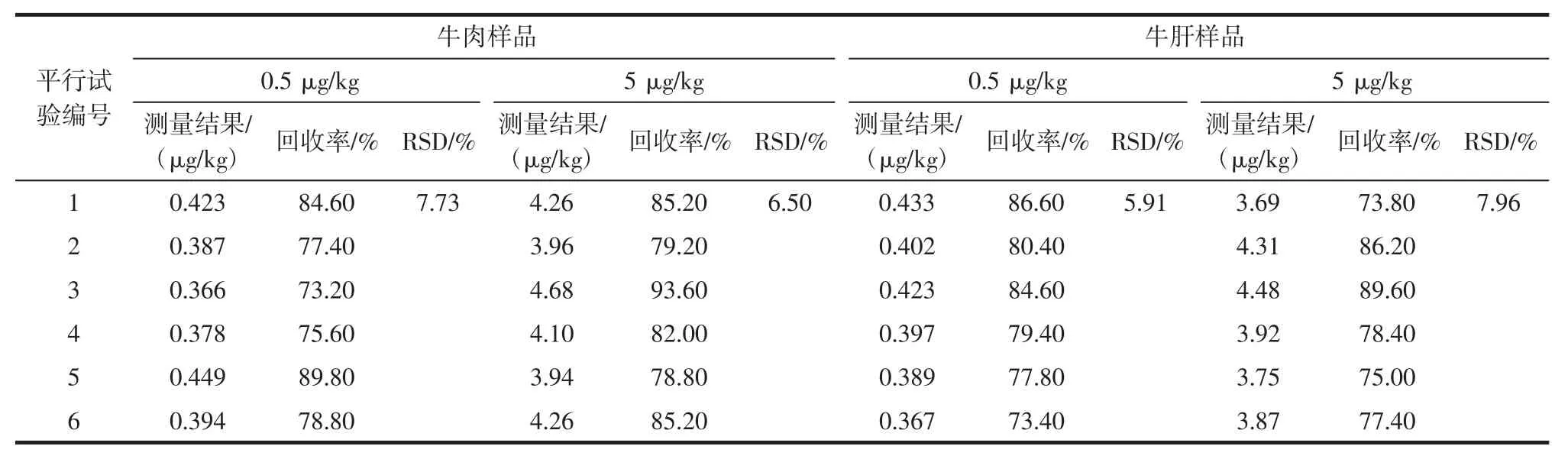
表3 牛组织中常山酮的回收率和精密度结果Table 3 The standard recovery and precision of halofuginone in bovine tissues
3 结论
本试验建立了牛组织中常山酮残留量的超高效液相色谱-串联质谱检测方法,样品经胰蛋白酶酶解,乙酸乙酯提取,固相萃取柱净化,液相色谱-串联质谱法测定。本方法具有检测快速、灵敏度高、定性定量准确、杂峰干扰小、检出限低等特点,适用于牛组织中常山酮残留量的准确定性和定量。
